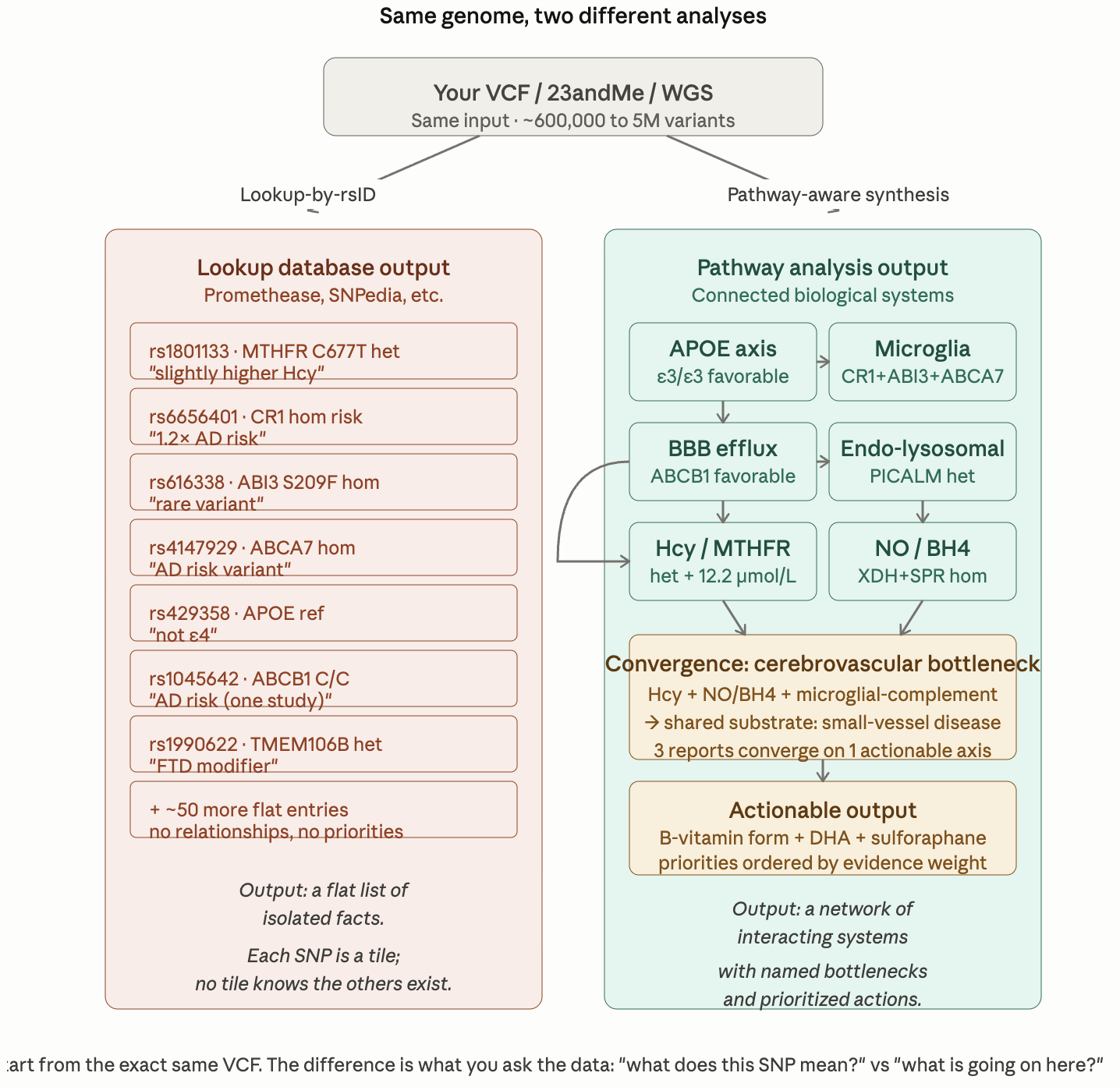
Here is the comparison between what I do in those deep dive reports vs simple database lookups like Promethease, SNPedia, FoundMyFitness Genome, and similar tools.
What lookup databases (Promethease) give you
Promethease, SNPedia, FoundMyFitness Genome, and similar tools work the same way: they take your VCF, match each rsID against a curated database of published associations, and produce a sortable list. For each variant they show what the literature says — odds ratio, study citation, and a one-line interpretation. The tools are useful for what they are: a fast, comprehensive, automated literature lookup.
The output is a flat catalogue. Each entry is independent. There are typically 20,000 to 60,000 reportable variants for a person, and the user is expected to filter, prioritize, and integrate them by hand. The tool does not know that rs1801133 (MTHFR) and rs6656401 (CR1) are biologically related, because no database edge connects them. They sit in separate rows, evaluated separately.
The diagram makes the structural argument visible. Below is the explanation that goes with it — the four things a pathway analysis does that a lookup database structurally cannot.
What lookup databases give you
Promethease, SNPedia, FoundMyFitness Genome, and similar tools work the same way: they take your VCF, match each rsID against a curated database of published associations, and produce a sortable list. For each variant they show what the literature says — odds ratio, study citation, and a one-line interpretation. The tools are useful for what they are: a fast, comprehensive, automated literature lookup.
The output is a flat catalogue. Each entry is independent. There are typically 20,000 to 60,000 reportable variants for a person, and the user is expected to filter, prioritize, and integrate them by hand. The tool does not know that rs1801133 (MTHFR) and rs6656401 (CR1) are biologically related, because no database edge connects them. They sit in separate rows, evaluated separately.
What the pathway approach does that the lookup approach cannot
1. It evaluates genes in the context of the system they belong to, not in isolation
A SNP is meaningful only relative to the pathway it sits in. CR1 homozygous risk is interesting on its own; CR1 homozygous risk plus ABI3 homozygous plus ABCA7 homozygous plus MS4A heterozygous burden is a coherent microglial-complement bottleneck — same biological function, four genes, mutually reinforcing. The lookup database lists the four entries on four different rows and lets the user notice the pattern. The pathway approach names the bottleneck, explains the mechanism, and lists the genes that participate in it as a single unit.
This is the difference between a glossary and a textbook. The glossary has every term defined correctly; the textbook tells you which terms belong in the same chapter and why.
2. It detects convergence across pathways — which is where the biology actually lives
The lookup database evaluates genes one at a time, so it cannot see that MTHFR het (homocysteine pathway) + XDH/SPR/GCH1 (NO/BH4 endothelial pathway) + CR1/ABI3/ABCA7 (microglial pathway) all converge on the same downstream organ — the cerebrovascular endothelium and small vessels of the brain. Three independent genetic findings, three different reports, but one shared bottleneck. That shared bottleneck is what determines the actual clinical priority, and it cannot be extracted from any single SNP entry.
The same convergence logic surfaces other patterns the same way: glucose / glycation / advanced glycation end products converging on diabetic-retinopathy-like microvascular biology; rapamycin’s IGF-1 longevity benefit converging with autophagy-mediated Aβ clearance from a completely different report. These connections are invisible to a flat-list approach.
3. It interprets variants against a person’s specific biological background, not against population averages
The same SNP can mean opposite things in different people. ABCB1 C3435T C/C is called “AD-risk” in the Cascorbi 2014 candidate-gene literature and “favorable for BBB Aβ efflux capacity” in the BBB-PET pharmacology literature. A lookup database cites both findings as separate independent entries and leaves the user to reconcile them. The pathway approach reads the call against the rest of the genome — APOE genotype, microglial findings, complement axis — and explains which interpretation is more likely to apply to this person’s biology.
This is also where the APOE keystone matters most. Whether a finding is “moderate concern” or “negligible” frequently depends on APOE genotype: an ε4 carrier has a fundamentally different risk landscape than an ε3/ε3 carrier, and the same downstream variant (CR1 hom, TREM2 R47H, or anything microglial) carries different weight against each backdrop. A lookup tool cannot adjust for this; the SNP entry is the same regardless of who is looking.
4. It links genetics to actionable inputs, ordered by evidence weight
The lookup database tells the user “you have rs1801133.” The pathway approach tells them: this gene sits in the one-carbon methylation pathway, the cofactors are 5-MTHF / methyl-B12 / P5P / B2 / TMG, the relevant biomarker is plasma homocysteine, the evidence base spans Casas 2005 Mendelian randomization for stroke and Wang 2022 BMJ for dementia, and the practical input is verifying the methyl-B12 form in the patient’s existing multivitamin. The biology connects the SNP to a specific intervention through a named cofactor, with citations. The lookup tool stops at the SNP.
This is also why the pathway approach can flag what’s missing. A flat list cannot say “you should consider sulforaphane” because it has no concept of biological gaps. The pathway approach can name a cofactor (NRF2 activation), notice that no current supplement covers it, see that five other reports independently flag the same NRF2 axis, and prioritize the addition. That recommendation cannot fall out of a per-SNP lookup at any level of completeness.
What the lookup approach is genuinely better at
Lookup databases are faster, more comprehensive in literature coverage, and easier to keep up to date. They surface unusual rare variants the pathway author may not have catalogued. They give a starting list of every reported association in the literature without requiring the analyst to pre-decide which pathways to characterize. They are cheap, automated, and reproducible.
The right way to use them is as a complementary first pass: scan the lookup output for anything unexpected, then run the pathway analysis to interpret the findings in context. The pathway analysis does not replace the lookup — it does a different thing. The lookup tells you what is in the genome; the pathway analysis tells you what is going on in the organism.
The one-line summary
A lookup database answers “what does this SNP mean?” — one question, asked thousands of times in parallel. A pathway analysis answers “what is going on here?” — a single question that requires every relevant SNP, biomarker, environmental exposure, and current intervention to be considered jointly. Most clinically meaningful patterns in a genome are visible only to the second question.