Of course, we all make decisions under uncertainty. The evidence in favor of B12 supplementation in people without B12 deficiency to prevent PD is just not there.
1 Like
@adssx 's point is that people should evidence their argument rather than “make a leap of faith”.
B6, 9 and 12 are interesting in lots of ways. However, B6 particularly has a u shaped curve because of self-inhibition, B9 personally I think the normal range may be actually below optimal values.
B12 I don’t think have evidence for oversupplementation.
1 Like
I never said there was any benefit for PD. All my post were off-topic. Maybe that’s where the misunderstanding came from.
SGTL2i seem like a no brainer to me for PD: Canagliflozin - Another Top Longevity Drug - #1742 by adssx
4 Likes
Keeping glucose low strikes me as a good strategy. Avoid glucose spikes.
3 Likes
SGLT2i are also mild brain-penetrant mTOR inhibitors (indirectly?):
- Canagliflozin - Another Top Longevity Drug - #1724 by Davin8r
- Canagliflozin - Another Top Longevity Drug - #1543 by adssx
- Canagliflozin - Another Top Longevity Drug - #1466 by adssx
- Canagliflozin - Another Top Longevity Drug - #572 by adssx (intermittent?)
- Canagliflozin - Another Top Longevity Drug - #438 by adssx (“selective inhibition of cardiac mTORC1 but the concomitant activation of mTORC2”)
2 Likes
Combination therapies: a change in approach for Parkinson’s research
Therefore, we aim to encourage researchers to think about, and test, drug combinations earlier in the drug discovery process; Cure Parkinson’s are championing the testing of combination therapies with the hope of increasing the odds of identifying disease-modifying therapies with urgency.
To kick start this change, Cure Parkinson’s will be opening a £2 million funding call in October 2025 to encourage the testing of combination therapies for Parkinson’s.
@John_Hemming: which combinations would you like to see tested? Citrate + melatonin + K2 I guess?
1 Like
That would be a good start, but these things have important aspects relating to dosing and timing of dose. I don’t think CP would want to try 1g of melatonin at night, for example, or even 100mg.
It is one really which requires individual experimentation with potentially quite substantial doses
Well, that really opens up possibilities. In particular, combining drugs where one or both individually may have failed, because the combination is a whole new effect - we’ve seen this over and over again, like the mice lifespan studies I discussed recently - metformin alone failed but in combo with rapa worked, or simvastatin and ramipril both failed individually, but gave a good 9% extension combined.
There were several very promising drugs which we had high hopes for PD for mechanistic reasons, but which sadly and disappointingly failed trials. Combining them might give them a new lease on life. I think that’s a very exciting possibility. We might be giving up on some drugs too soon!
2 Likes
Already posted: Parkinson's disease - #885 by adssx
However… What do we do with that post infection? Immuno modularity drugs?
If a virus damages the mitochondria the mitochondria need to be repaired. If the mitochondria have not yet been repaired then a drug to fight the virus may be some use.
2 Likes
Forgot to cross-post this great paper here:
- Oxygen, hypoxia and hyperoxia - #202 by J0hn
- Oxygen, hypoxia and hyperoxia - #203 by adssx
- Oxygen, hypoxia and hyperoxia - #206 by adssx
And new tiny RCT (n=2): Effects of Intermittent Hypoxia Training with Parkinson’s Disease 2025
Purpose: This case study investigated the effects of intermittent hypoxia training (IHT) on dopamine (DA) levels in an individual with Parkinson’s disease (PD), compared to a healthy control. The aim was to assess the feasibility, safety, and potential dopaminergic response to a six-week IHT intervention. Methods: Two female participants (one diagnosed with PD and one age-matched control) completed twice-weekly sessions of low-intensity cycling in a normobaric hypoxia chamber (FiO₂ 14.9%) for six weeks. Peripheral oxygen saturation was monitored to ensure safety, and venous blood samples were collected at baseline, midpoint (3 weeks), and endpoint (6 weeks) to evaluate serum dopamine levels using a competitive ELISA. Results: The control participant demonstrated a progressive increase in DA concentration, reaching a 104.9% increase by the study endpoint. The PD participant also showed elevated DA levels, with a 39.3% increase from baseline. The intervention was well tolerated by both participants, with no adverse events reported. Conclusion: Preliminary findings suggest that IHT may elicit a dopaminergic response in individuals with PD, potentially supporting its use as a novel adjunctive strategy to enhance neuroplasticity and dopaminergic activity. These results support the feasibility and safety of IHT in a clinical context; however, further investigation in larger cohorts is required to determine efficacy.
Intermittent hypoxic exercise was conducted using a normobaric hypoxia chamber fitted by the Altitude Centre (Bank, United Kingdom) at the East London University, UK. Sessions were calibrated to an oxygen concentration of 14.9%. Each training session consisted of five minutes of continuous cycling inside the hypoxia chamber, followed by two minutes of seated rest in normoxia conditions. Two participants (n = 2) cycled at a cadence of 60 revolutions per minute (rpm) on a stationary Monk exercise bike without additional resistance, maintaining a light-intensity workload.
Peripheral oxygen saturation (SpO₂) was continuously monitored using a handheld pulse oximeter (MD300M, ChoiceMMed, Beijing, China) to ensure values did not fall below 86% during exposure
The intervention was carried out twice weekly (Fridays and Saturdays) for a total duration of six weeks, at the same time of day for each session to control for circadian variation. Participants were advised to abstain from physical activity for at least 24 hours and from stimulants (e.g., caffeine) for 48 hours prior to each session.
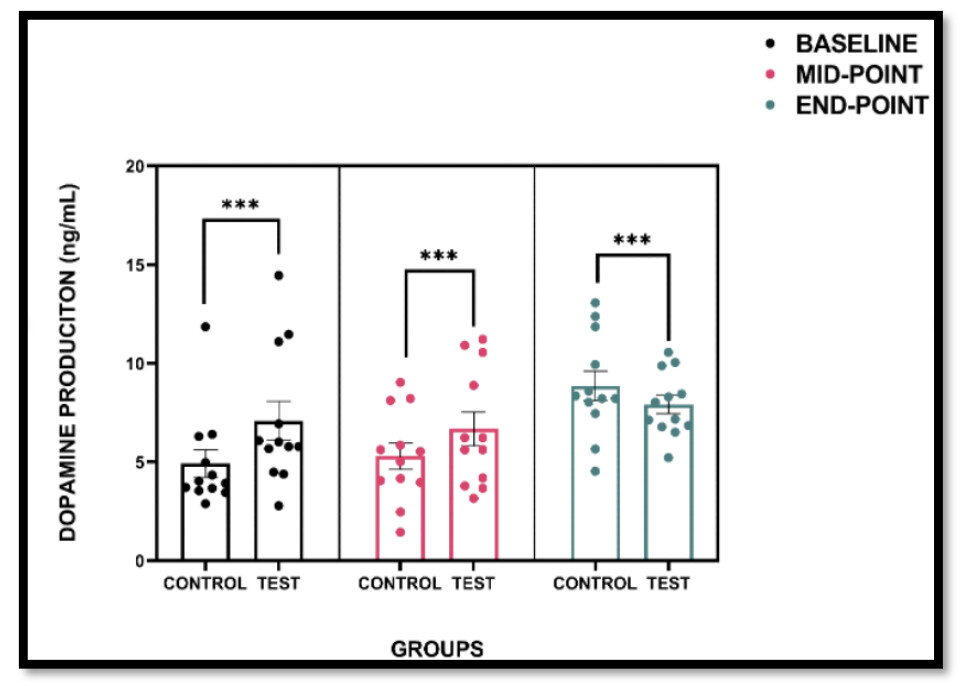
Not very clear to me what this graph shows and the way the graph is described in the paper is contradictory.
Emerging evidence suggests that the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection may have long-term deleterious effects on the central nervous system and even contribute to post-COVID neurological syndromes. Interestingly, inflammation-induced proteolytic processing of the Spike protein of SARS-CoV-2 leads to the generation of peptides capable of aggregating into amyloid fibrils in vitro. Herein, we investigate the in vitro effect of a fibrillogenic fragment of the Spike protein [Spike 194–203 (S194)] on the aggregation and toxicity of the Parkinson’s disease (PD) protein α-synuclein (αSyn). Our results indicate that S194 fibrils stimulate in a concentration-dependent manner the fibrillation of αSyn monomer, resulting in aggregates with increased capacity of inducing lipid vesicle leakage and toxicity to neuroblastoma cells, in comparison with either αSyn or S194 alone. Bidimensional NMR (1H–15N-HSQC) suggests that S194 fibrils cause a higher perturbation in both the N-terminal region (sequence: 19–68) and the hydrophobic central domain of the αSyn monomer (sequence: 71–95), which is corroborated by protein–peptide docking and molecular dynamics simulations. In contrast with fibrils from wild-type αSyn, aggregates from the PD variant A30P exhibited a remarkable accelerative effect on S194 fibrillation. Similarly, fibrils from amyloid-β peptides, which are linked to Alzheimer’s disease, exhibited a pro-aggregating effect on the S194 monomer. Taken together, these findings might contribute to a broader understanding of the potential connections between SARS-CoV-2 infection and amyloid-related neurodegenerative disorders, highlighting areas that may warrant further investigation.
3 Likes
Related: Cerebellum mitochondrial DNA copy number is increased in Parkinson’s disease 2025
Bioinformatics methods can be used to quantify mitochondrial DNA copy number from whole genome sequencing (WGS) data. We evaluated mitochondrial DNA copy number from human brain-derived WGS data using the fastMitoCalc tool. 341 Parkinson’s Disease cerebellum samples were compared with 74 age-matched controls from the North American Brain Expression Consortium. Parkinson’s Disease cerebellum had significantly higher mitochondrial DNA copy number compared with controls (P = 4.15e–7), and this effect was reproducible in four of five brain banks when analysis was restricted to each resource that contributed Parkinson’s Disease samples to this genetic dataset. Follow-on analyses of 128 Parkinson’s Disease cerebellum samples and 33 controls that had paired neuropathology data and clinical scores demonstrated a significant increase in mitochondrial DNA copy number with Unified Staging System for Lewy Body disorders stages and Unified Parkinson’s Disease Rating Scale (off meds) motor scores. Analysis of Lewy Body scores from ten brain regions showed cerebellum mitochondrial DNA copy number increased upon pathological infestation of α-synuclein aggregates in the brainstem and limbic system but did not increase after late-stage neocortical involvement. This genetics dataset supports previous observations of cerebellum activation in Parkinson’s Disease and suggests mitochondrial DNA copy number may increase to support this regional activation as a compensatory mechanism to pathology or motor symptoms.
That could be because of deletions. Shorter mtDNA has a replication advantage. That does not make it any better and in fact it is worse.
Are you saying that there are more deletions in PD and cells with mtDNA deletions compensate by increasing the DNA copy number? Or did I misunderstand completely?
At an early stage PD has more deletions. At a later stage the cells with deletions have stopped functioning.
https://www.thelancet.com/pdfs/journals/ebiom/PIIS2352-3964(24)00100-2.pdf
Findings Patients with IRBD, both converters and non-converters, exhibited more cf-mtDNA with deletions in the CSF than controls. This finding was confirmed in CD9-EVs. The high levels of deleted cf-mtDNA in CSF corresponded to a significant decrease in cf-mtDNA copies in CD9-EVs in both IRBD non-converters and converters. Conversely, a significant increase in cf-mtDNA copies was found in serum and CD9-EVs from the serum of patients with IRBD who later converted to a Lewy body disorder.
Interpretation Alterations in cf-mtDNA copy number and deletion ratio known to occur in Lewy body disorders are already present in IRBD and are not a consequence of Lewy body disease conversion. This suggests that mtDNA
dysfunction is a primary molecular mechanism of the pathophysiological cascade that precedes the full clinical motor and cognitive manifestation of Lewy body disorders.
Deletions have a replicative advantage because they result in shorter DNA. (particularly the common deletion).
Chat GPT has given me this list of papers to look at, but I have not done so as yet.
-
2022 — Cell-free mtDNA deletions in iPD vs LRRK2-PD (CSF, multiplex dPCR):
https://pubmed.ncbi.nlm.nih.gov/36208866/ -
2024 — mtDNA deletions in CSF of idiopathic REM sleep behaviour disorder (prodromal Lewy body disease):
https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(24)00100-2/fulltext
(PDF: https://www.thelancet.com/pdfs/journals/ebiom/PIIS2352-3964(24)00100-2.pdf ) -
2024 — Plasma cf-mtDNA deletion level in PD & MSA (qPCR/ND1–ND4 proxy):
https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2024.1488820/full
(PMC mirror: https://pmc.ncbi.nlm.nih.gov/articles/PMC11647036/ ) -
2023 — mtDNA damage/deletions trigger spread of PD pathology (mouse & human context):
https://www.nature.com/articles/s41380-023-02251-4 -
2024 — Methods: “Common mitochondrial deletions in RNA-Seq” (bulk, single-cell & spatial):
https://www.nature.com/articles/s42003-024-05877-4 -
2025 — Review: “Mitochondrial DNA (mtDNA) as fluid biomarker in neurodegenerative diseases”:
https://pmc.ncbi.nlm.nih.gov/articles/PMC11744304/ -
(Alt open-access mirror for the iRBD CSF paper):
https://pmc.ncbi.nlm.nih.gov/articles/PMC10963194/
I’ve been looking at passion fruit extracts for AD prevention and generally neuroprotection, and incidentally came across some interaction with PD. There are many species of these plants and different extracts, so that’s all over the place. Plus these are in mice with induced PD (shit model), so massive caveats, but just in case this might be of interest, I thought I’d post it. Nothing to get excited over, I came across this incidentally, but anyway.
Passiflora cincinnata Extract Delays the Development of Motor Signs and Prevents Dopaminergic Loss in a Mice Model of Parkinson’s Disease
https://onlinelibrary.wiley.com/doi/10.1155/2017/8429290
Or: PMC5556616
The neuroprotective properties of a novel variety of passion fruit
https://www.sciencedirect.com/science/article/abs/pii/S1756464616000955
1 Like