Metabolic Dysregulation in Parkinson’s Disease: Non-Oxidative Phosphorylation and Its Role in Brain Energy Metabolism 2025
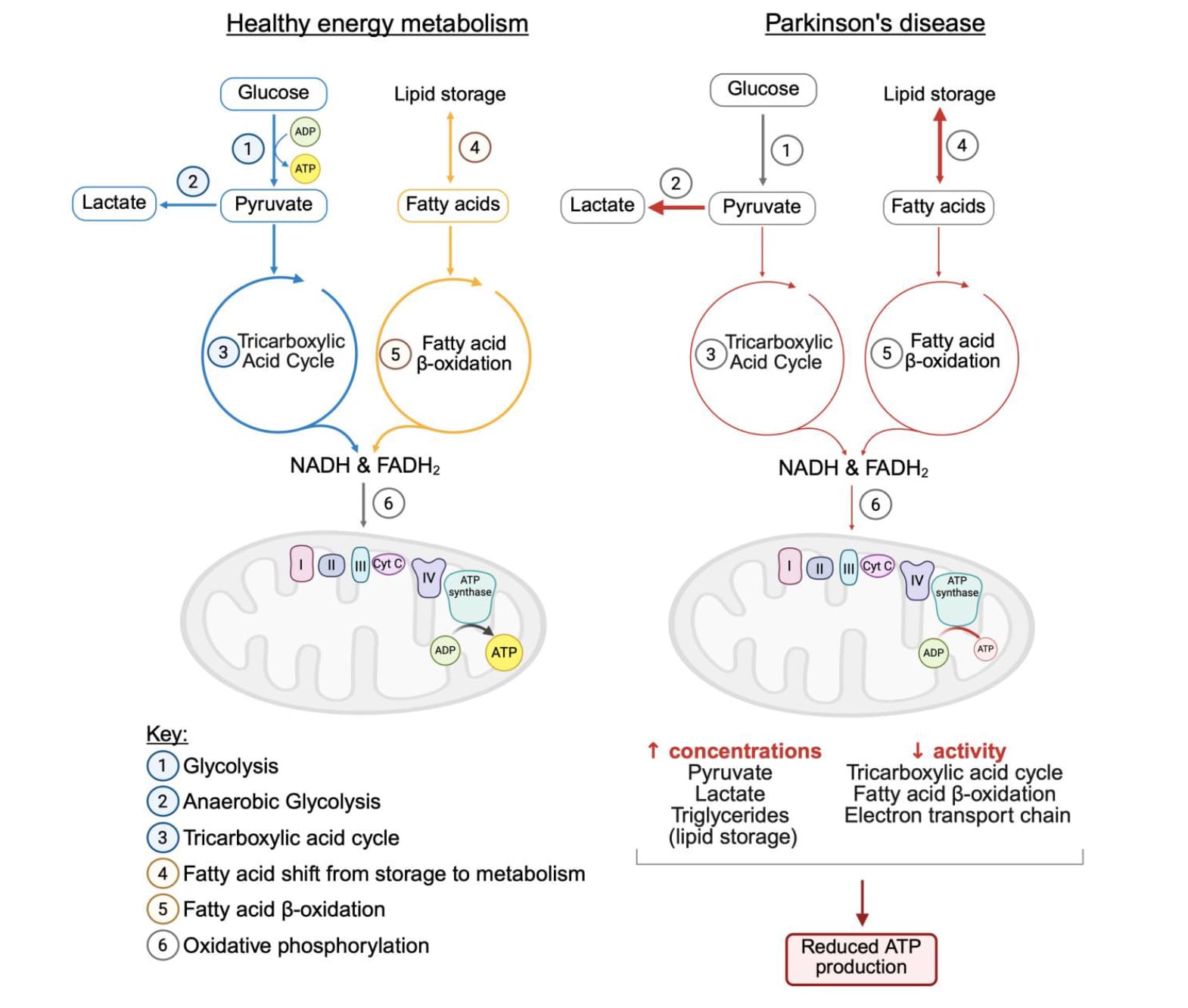
Parkinson’s disease (PD) is a progressive neurodegenerative condition affecting around 1-2% of the population over the age of 60. The lack of disease-modifying therapies highlights the need for insights into the etiology and pathogenesis of PD. Mitochondrial dysfunction is recognized to be a significant contributor to disease pathogenesis, resulting in bioenergetic deficits and subsequent neurodegeneration. Research indicates that changes in non-oxidative phosphorylation (non-OXPHOS) metabolism in PD may serve as an adaptive response to mitochondrial dysfunction, compensating for energetic failure and alleviating disease progression. This review explores mitochondrial dysfunction-driven alterations in non-OXPHOS metabolic pathways, such as glycolysis and the tricarboxylic acid cycle, emphasizing their role in maintaining energy metabolism and their dual contribution to neuroprotection and disease progression. Advances in neuroimaging techniques are also discussed, particularly their role in visualizing metabolic changes in vivo and their potential utility in identifying non-OXPHOS metabolism as a biomarker of mitochondrial dysfunction. By enhancing our understanding of the complex interplay between metabolic pathways in PD, this review underscores the importance of personalized therapeutic approaches that consider individual metabolic variations. Ultimately, these insights aim to pave the way for improved diagnostic utility and personalized treatment strategies that address the metabolic and mitochondrial dysfunctions underlying PD pathogenesis.
Full paper: PDF
@John_Hemming: non-OXPHOS and TCA cycle 
On glycolysis: “The evidence of glycolysis’ role in PD is conflicting, demonstrating the upregulation of specific glycolytic enzymes as a neuroprotective mechanism, while increased glycolytic flux results in the accumulation of toxic end-products, exacerbating neurodegeneration. The dual nature of glycolysis necessitates identifying the threshold balancing the compensatory benefits against neurotoxic effects. The lack of consistency suggests that the glycolytic reprogramming in PD is context-dependent, with factors such as cell type, disease stage, underlying genotype, and threshold of glycolytic flux shaping its contribution to disease progression.”
On TCA cycle: "This process is significantly disrupted in PD, as evidenced by findings from in vivo PD models. PDH catalyzes the first gatekeeping step of the TCA; however, its activity is compromised in PD. […] The consistent evidence of reduced PDH activity suggests TCA cycle decoupling from glycolysis, leading to elevated pyruvate levels and reduced glucose flux through oxidative metabolism. Elevated pyruvate levels are converted to lactate due to increased LDH activity in PD, signifying a metabolic shift towards anaerobic glycolysis. […] MPP±treated SH-SY5Y cells demonstrated reduced citrate synthase (CS) and isocitrate dehydrogenase activity (IDH)
Fatty acid β-oxidation: “Similarly to glycolysis, FAO appears to be differentially affected across disease stages – upregulated in early stages to meet energy demands and declining with the disease progression; however, this pattern remains controversial and not fully supported by all research available. […] Evidence linking dysregulated FAO to pathogenesis is increasing; for instance, protein analysis of post-mortem tissue revealed early-stage FAO upregulation, marked by increased expression of acetoacetyl-CoA thiolase, a key FAO enzyme [37]. Reduced mitochondrial complex I activity correlated with abnormal FAO, marked by increased plasma levels of isobutyrylcarnitine, reinforcing FAO dysregulation as a consequence of mitochondrial dysfunction [38]. Despite the evidence from some studies reporting FAO upregulation, its overall function is disrupted, as indicated by a shift from FA metabolism to lipid storage in PD.”