Which is why there’s a search for biomarkers which could be proxies for the process. If we establish firmly that, say, ApoB levels at given values “guarantee” no atherosclerosis, then all we have to show is that a given drug brings ApoB levels to that given value and we don’t have to wait for 20 years to show atherosclerosis prevention. Obviously, that has all the disadvantages of biomarkers as proxies, questions of drug tolerability and long term side effects etc., but you release the drug into the wild and wait for the clinical data to gather over the years (statins).
If amyloid and tau tangles prevention can definitively guarantee no ND disease (say, AD), then perhaps some biomarker(s) can be a proxy. Then you show that the drug candidate affects the biomarker etc.
What biomarkers or measurements can act as such proxies for any given NDD?
How can we measure aging? If a given drug (say, rapamycin) affects those key markers then we don’t have to wait a whole human lifespan to assess effectiveness, because we can show impact on those proxies. We first validate those proxies in animal models, and then see if there’s a similar effect in humans. That’s what Fontana et al tried to do in the CALERIE study for the effects of CR in humans. We have a bunch of biomarkers associated with CR in animal models. If we can now show that initiating CR in humans gives us a similar biomarker profile as in animal model CR, then we have a leap of faith that there will be a similar longevity outcome. It’s the best we can do, because nobody is doing a lifelong controlled study in humans. On the other hand, we have the Framington multigenerational CVD data etc., so observational approaches have been used with decent confidence - based on this, recommendations have been generated. Maybe something similar could be tried.
You start with animal models. First validate it there, then do trials of the candidates in humans. We don’t have good animal models for AD to begin with, the mice models we are using are shit. Even then, when it comes to amyloid it’s chicken and egg.
Chinese, but Beijing:
Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives
Your last statement is exactly why I assume there’s skepticism of getting a drug proven to improve cognitive decline by 75%: not the science, but the logistics, incentives, and timelines. " it’s a problem similar to rapamycin and all longevity trials" — and ACM for statins, as I pointed out.
However, I do think we will get this answer eventually, and in the positive. In 3.5 years, I expect that they will be able to prove that they can clear amyloid beta out of the brains of cognitively normal people with minimal side-effects. Then they can observe them without further intervention (or, at their option, invite them for further treatment on an open-label basis, possibly on a less frequent dosing schedule since they’ve already roto-rootered their brains) for 5-15 years more and see if it substantially reduces the rate at which people transition into MCI or dementia, which (a) would likely allow them a license extension, and (b) might already qualify as a 75% reduction in cognitive decline and certainly would if sustained for another few years.
As CronosTempi notes, they might try for a conditional license extension earlier on the basis of the biomarker change plus safety, analogous to what was previously done with Aduhelm but on more solid footing since their Phase 3s unambiguously succeeded. (CT: Aduhelm was approved in substantial part on the basis of having reduced brain amyloid beta. We now have additional, well-validated serum markers, such as ptau-217).
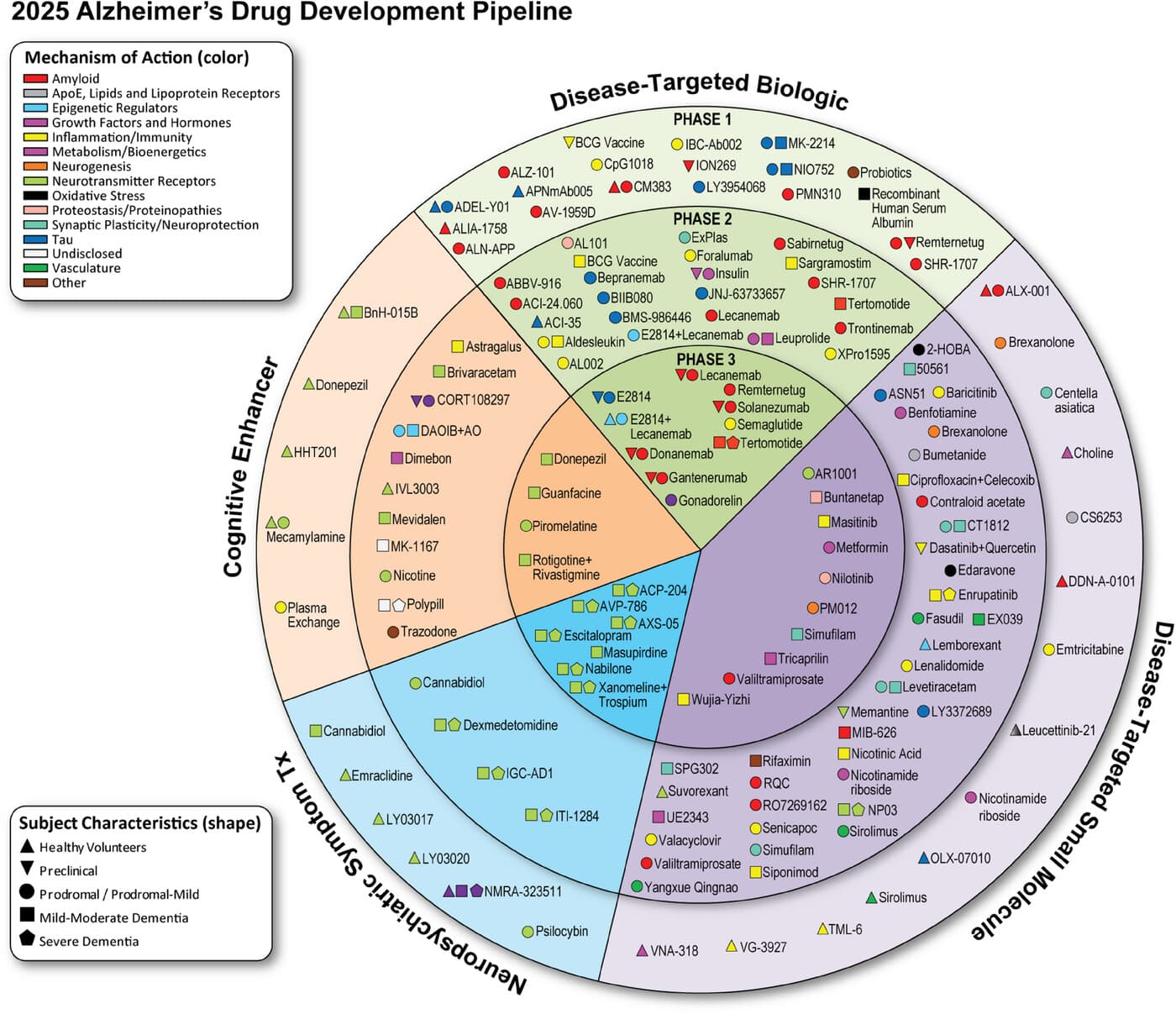
Aducanumab was abandoned. We still have donanemab and lecanemab. They’re doing what you’re suggesting:
NCT04437511: “Following the double-blind 76-week main study period, a double-blind 78-week long-term extension period is added to further evaluate donanemab efficacy and safety over time.”
NCT05026866: Donanemab: “Approximately 800 additional participants will be enrolled in a 12-month addendum to assess safety of a different titration regimen.” (that’s the one cited by the author when he writes: “On the other hand, if a clinical trial completes earlier than 12 years from now (perhaps [73], reading out in 2027), sustains extremely good amyloid clearance at the preclinical stage, and has a good safety profile, but doesn’t make substantial progress towards this 75% goal, then I would consider this prediction refuted in advance.”)
NCT05508789: “The study duration including screening and follow-up is up to 93 weeks.” in “prodromal AD and mild dementia due to AD”
NCT06566170: “The study will last about 273 weeks and may include up to 28 visits.” (5 years!)
NCT06889818: 10y follow up
NCT03887455: Lecanemab, 69 months (almost 6y)
NCT04468659: Lecanemab, 216 weeks (4y)
These drugs clear amyloid perfectly, yet prediction markets are skeptical about their effectiveness: why?
My other concern is that even if we manage to prove that taking these drugs early slows down cognition decline by >75%, what about other effects? If they increase ACM, then who would take them? If they decrease ACM then they would be amazing longevity drugs but we would see lifespan extension in mice if that was the case. That’s why targeting tau might be more promising, even if more downstream than amyloid: you can intervene later and act when people are already somewhat impaired so that you improve their quality of life and ACM matters less. (And it costs less to prove the efficiency with shorter trials…)
From the summaries you gave, none of these studies are doing what I’m suggesting (though I expect some of them will if they’re successful as I defined above). Most of of these studies last three years or less. NCT06566170 is in “Participants With Early Symptomatic Alzheimer’s Disease”. NCT06889818 is an observational study, not a trial, and is in people with AD. NCT03887455 is the extension study of the already-completed Phase 3 for Lequembi, whose subjects had AD. NCT04468659 is the right kind of study, but only 4 y (though I expect they will ultimately do what I suggest, if successful as I defined above).
To avoid AD, I actually expect a lot of people would take them. But in any case, there’s no reason to think they would do this.
… if anyone had ever done a lifespan study with them, which no one has …
Actually, you wouldn’t expect a LS increase from these drugs in wild-type mice, because mice don’t naturally accumulate amyloid beta in their brains. You could do it in AD-transgenic mice, but I don’t think many people would take a “life extension” in such mice seriously (and they shouldn’t).
I do think it’s causal — newborn infants have apoB, but it’s still causal for ASCVD (yes, I know their levels are low) — but it doesn’t matter whether it’s causal — just that it’s predictive as a biomarker, which it is.
But often on top of previous studies of ~2y so aligned with the 5y goal you mentioned.
Aren’t there long-term carcinogenicity studies in mice before approval? I remember those for dapagliflozin and empagliflozin that we discussed in the canagliflozin thread. Not a lifespan study but gives some hint.
None of the relevant studies as defined are on top of anything. The studies in cognitively-normal people positive for beta-amyloid are only now starting from scratch. And I suggested we would get a positive result if a 3.5 year trial were followed by an additional 5-15 years of open label, so that’s 8.5-18.5 years, not 5, 3, or 2.
“Gives some hint” in a 2-y carcinogenicity study is not “are amazing longevity drugs” as you proposed in the negative, arguing from silence. And an antibody designed to fix a problem that wild-type mice don’t have could conceivably even be slightly harmful to them but positive for ACM in humans.
As I said, that kind of concern “is exactly why I assume there’s skepticism of getting a drug proven to improve cognitive decline by 75%: not the science, but the logistics, incentives, and timelines.” But I do anticipate we’ll get the data.
I will note that Tanzi is one of the most prominent advocates of an infectious hypothesis for AD — and in his view, it goes through amyloid beta.
No new worrisome safety signals have appeared; rates of the treatments’ most serious adverse event have not exceeded those seen in clinical trials. “If there was a really bad safety signal, we would have seen it,” noted clinical trialist Robert Howard, MD, a professor of old age psychiatry at University College London. “Give the drugs credit where it’s due. That’s good news.”
Eisai presented data from its 48-month follow-up at this year’s AAIC. However, the company this year compared changes in cognition and function over time, again as determined by the CDR-SB, only between trial participants who received lecanemab from the start and 2 historical control groups. The delayed-start trial participants were not included in the comparison. Howard said he can’t help but wonder whether Eisai this year decided to omit the delayed-start patients from the comparison because they had caught up with the early-start patients, which would suggest that lecanemab wasn’t a disease-modifying therapy after all.
In an email, Eisai spokeswoman Libby Holman said that the company had not planned to compare the early-start vs delayed-start trial participants past 36 months.
Their manufacturers did not provide uptake data on the therapies requested by JAMA Medical News. University College London’s Howard says his sources tell him “the numbers are really, really small,” with only around 13 500 patients worldwide having been treated with lecanemab.
Whether the treatments actually extend the time patients can run errands or play with their grandchildren—the kinds of benefits that Pike of the Alzheimer’s Association referred to when the FDA granted traditional approval to lecanemab—can’t be determined in clinical practice because there isn’t a placebo comparison group, University College London’s Howard pointed out. “There is a massive placebo effect.”
Just as the safety profiles of the antiamyloid therapies in the real world appear to be about the same as in their clinical trials, Howard said, “the drugs are probably only as effective as they were in the trials, and that isn’t very effective.”
Colleagues in Japan and South Korea who have treated patients with the drugs have told him that “all we’re seeing is people deteriorating the way they do without treatment,” Howard said.
Whether amyloid-β (Aβ) peptides are synaptogenic or synaptotoxic remains a pivotal open question in Alzheimer’s disease research. Here, we chronically treated human neurons with precisely controlled concentrations of chemically defined synthetic Aβ40, Aβ42, and Aβ42arctic peptides that exhibit distinct aggregation propensities. Remarkably, chronic exposure of human neurons to free Aβ40 at higher concentrations or to free Aβ42 at lower concentrations potently promoted synapse formation. In contrast, aggregated Aβ42 or Aβ42arctic at higher concentrations were neurotoxic and synaptotoxic. The synaptotoxic effects of Aβ peptides manifested as an initial contraction of the synaptic vesicle cluster followed by synapse loss. Aβ40 and Aβ42 peptides with scrambled or inverted sequences were inactive. Thus, our experiments reveal that Aβ peptides exhibit an aggregation-dependent functional dichotomy that renders them either synaptogenic or synaptotoxic, thereby providing insight into how Aβ peptides straddle a thin line between physiological synapse organization and pathological synapse disruption. Among others, our data suggest that Alzheimer’s disease therapies might aim to shift the balance of Aβ peptides from the aggregated to the free state instead of suppressing all Aβ peptides.
Thanks a lot. You’re right, here are excerpts from the paper:
Note that the lack of a toxic effect by Ab40 does not imply that Aβ40 is generally not neurotoxic since it seems likely that it is the oligomeric species of all Aβ peptides that is toxic. Under our precisely controlled conditions, Aβ40 is probably not incubated for a sufficiently long time to form aggregates or the nucleating effect of Aβ42 is missing, thus rendering it non-toxic
Our fractionation experiments strengthened this conclusion: the pellet containing only large aggregates and the supernatant enriched in free Aβ peptides and smaller oligomers produced indistinguishable toxicity, suggesting that this toxicity is not caused by the free Aβ peptides or by large Aβ aggregates but by species in an intermediate state of aggregation
Therapeutically, our results bolster current strategies aimed at neutralizing aggregated Aβ, as pursued by many ongoing clinical trials (4-6). Yet, our results also suggest that simply clearing all Aβ from the brain may be insufficient as an AD therapy and could even be counterproductive. In clinical trials where the elimination of aggregated plaques was significant, cognitive decline persistently continued (4-6). At the same time, in these trials increased free Aβ42 levels in the CSF correlated with better therapeutic outcomes (17). Such findings echo our observations by underscoring a potential protective or supportive role for free Aβ. We thus hypothesize that Alzheimer’s disease pathology is propelled not merely by the presence of aggregates but also by a depletion of free Aβ that occurs when Aβ peptides become sequestered into increasingly larger aggregates (33). Accordingly, Alzheimer’s disease treatments may need to preserve the levels of free Aβ in patients to sustain the synaptogenic capacity of Ab in addition to combating Ab aggregates to limit the synaptotoxicity of Aβ. Current clinical findings lend support to this view. The approved Aβ immunotherapies (aducanumab, lecanemab, donanemab) (6, 34, 35) were designed to recognize aggregated forms and spare monomeric Aβ, consistent with our conclusion that neutralizing aggregates is desirable. Conversely, solanezumab, an antibody that preferentially bound monomeric Aβ, failed to yield cognitive benefit, a result that may reflect the unwanted removal of the very species needed for synaptic support