Article combining our favorite topics (mtDNA, hypoxia, and PD): Hypoxia and TTR dysregulation in astrocytes from Parkinson’s disease with a specific mitochondrial haplogroup: A single-cell analysis 2025
Mitochondrial DNA variants have been linked to cognitive progression in Parkinson’s disease; however, the mechanisms by which mitochondrial DNA variants or haplogroups contribute to this process remain unclear. In the present study, we analyzed single-nucleus RNA sequencing data from 241 post-mortem brain samples across five regions to investigate the dysregulatory mechanisms associated with mitochondrial DNA haplogroup H and haplogroups J, T, and U#.
Pathway analysis highlighted abnormal hypoxic and reactive oxygen species environments in astrocytes, whereas protein complex analysis revealed a consistent and significant elevation in ribosomal subunit complexes within the astrocyte subtypes.
Our findings suggest that mitochondrial DNA haplogroup H may drive Parkinson’s disease cognitive progression through aberrant TTR expression and a hypoxic environment.
Gut microbial production of imidazole propionate drives Parkinson’s pathologies 2025
Parkinson’s disease (PD) is characterized by the selective degeneration of midbrain dopaminergic neurons and aggregation of α-synuclein. Emerging evidence implicates the gut microbiome in PD, with microbial metabolites proposed as potential pathological mediators. However, the specific microbes and metabolites involved, and whether gut-derived metabolites can reach the brain to directly induce neurodegeneration, remain unclear. Here we show that elevated levels of Streptococcus mutans (S. mutans) and its enzyme urocanate reductase (UrdA), which produces imidazole propionate (ImP), in the gut microbiome of patients with PD, along with increased plasma ImP. Colonization of mice with S. mutans harboring UrdA or Escherichia coli expressing UrdA from S. mutans increases systemic and brain ImP levels, inducing PD-like symptoms including dopaminergic neuronal loss, astrogliosis, microgliosis, and motor impairment. Additionally, S. mutans exacerbates α-synuclein pathology in a mouse model. ImP administration alone recapitulates key PD features, supporting the UrdA–ImP axis as a microbial driver of PD pathology. Mechanistically, mTORC1 activation is crucial for both S. mutans- and ImP-induced PD pathology. Together, these findings identify microbial ImP, produced via UrdA, as a direct pathological mediator of the gut-brain axis in PD.
The interesting bit is the mechanism:
Gut-colonized S. mutans induces dopaminergic neurotoxicity and motor dysfunction via mTORC1 activation
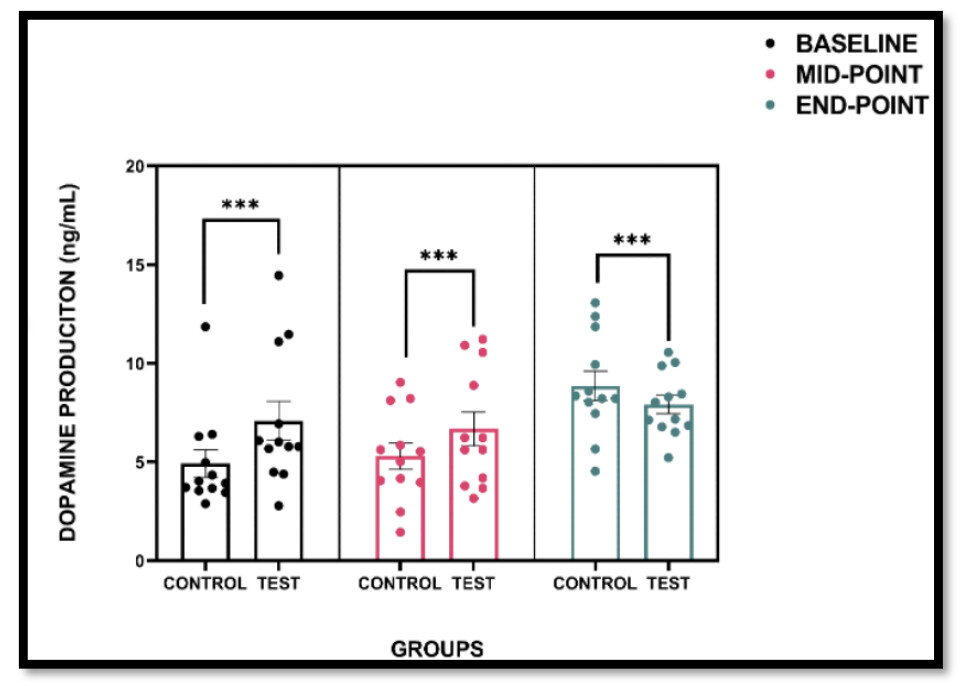
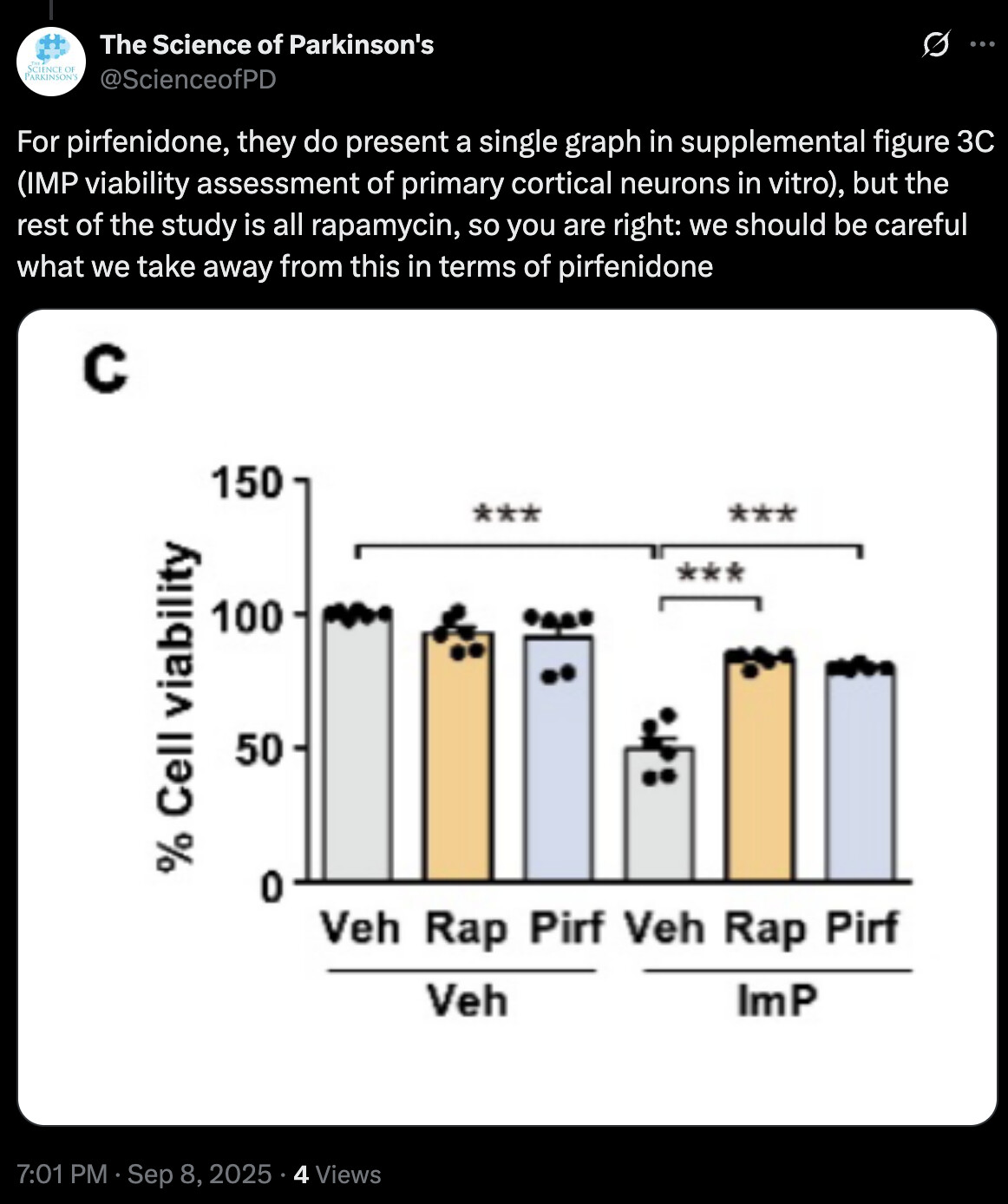
While imidazole propionate exerted relative regional specificity in inducing brain pathology in vivo, mTORC1 activation was still elicited in primary cortical neurons following imidazole propionate treatment in vitro, as evidenced by the phosphorylation of S6K1 at T389 and subsequent downstream signaling events, such as serine phosphorylation of IRS1 (Fig. 3c). These effects were effectively blocked by the mTORC1 inhibitor rapamycin or p38γ inhibitor pirfenidone (Fig. 3c), in line with the previous findings in primary hepatocytes9,14. Additionally, imidazole propionate treatment of primary cultured cortical neurons resulted in ~50.3% neurotoxicity, which was effectively blocked by both rapamycin and pirfenidone treatments (Supplementary Fig. 3c). These results underscored the key role of the imidazole propionate/p38γ/mTORC1 pathway in imidazole propionate-induced neuronal toxicity.
To investigate whether mTORC1 inhibition can reverse gut-colonized S. mutans-induced PD pathology, we depleted the gut microbiome of C57BL/6 N mice by administering an antibiotic cocktail (Abx) via oral gavage twice daily for seven days (Fig. 3d). This method, as described previously10,21, effectively reduces the bacterial load (Supplementary Fig. 3d), as monitored by a colony formation assay with fecal samples. Starting on the 8th day, the antibiotic-treated mice were gavaged with S. mutans (109 CFU/mouse) daily for 14 days with or without an intraperitoneal injection of rapamycin (Fig. 3d). S. mutans was undetectable before (Pre-Abx) and seven days after antibiotic treatment (Post-Abx), but its levels markedly increased following colonization. Rapamycin did not affect S. mutans colonization efficiency, as demonstrated by absolute quantification of the S. mutans 16S rRNA gene (Supplementary Fig. 3e). These treatments did not affect body weight or cecal weight (Supplementary Fig. 3f, g). Interestingly, S. mutans-induced decrease in brain weight in the antibiotic-treated mice was reversed by rapamycin treatment (Supplementary Fig. 3h). Although antibiotic-treated mice exhibited higher plasma and brain imidazole propionate levels than GF mice (Figs. 1j, k and 3e, f), S. mutans colonization further increased these levels to ~400 nM (Fig. 3e). Similarly, brain imidazole propionate concentration increased robustly in antibiotic-treated mice colonized with S. mutans (Fig. 3f). Importantly, rapamycin treatment did not reduce the elevated levels of imidazole propionate induced by S. mutans in plasma and brain (Fig. 3e, f). Precursor urocanate levels were similar among all experimental groups and were not affected by S. mutans colonization or rapamycin treatment (Supplementary Fig. 3i, j). The increase in S6 and 4E-BP1 phosphorylation induced by S. mutans in the dopaminergic neurons of the substantia nigra was almost completely reversed by rapamycin treatment (Fig. 3g, Supplementary Fig. 3k), despite comparable elevations in imidazole propionate in the brain of S. mutans-colonized mice with or without rapamycin (Fig. 3f). In support of our hypothesis of mTORC1-dependent neurotoxicity, rapamycin treatment (mTORC1 inhibition) effectively prevented 4E-BP1 phosphorylation, dopaminergic neurodegeneration, astrogliosis, and microgliosis in the ventral midbrain, as well as the loss of dopaminergic processes in the substantia nigra reticularis and dopaminergic axon terminals in the striatum (Fig. 3h, i, and Supplementary Fig. 3k, l, m). Rapamycin also reversed motor dysfunction induced by S. mutans colonization in antibiotic-treated mice (Fig. 3j).
Considering the increased risk of developing PD in individuals with diabetes and the association of this condition with increase in PD severity33, it is plausible that microbial imidazole propionate serves as a potential link between diabetes and PD. This hypothesis is based on our previous findings regarding its role in diabetes14. Based on the recent report demonstrating the interaction between p38γ and α-Syn and the translocation of p38γ to neuronal cell bodies in patients with dementia with Lewy bodies and PD34, the reversal of imidazole propionate-induced neurotoxicity by the p38γ inhibitor pirfenidone suggests a potential therapeutic option for individuals with elevated imidazole propionate levels and consequently high risks of diabetes and PD.
They write “the reversal of imidazole propionate-induced neurotoxicity by the p38γ inhibitor pirfenidone” but they only proved reversal with rapamycin. Weird. In any case @John_Hemming & @DrFraser it shows (again) that rapamycin could theoretically reverse some PD symptoms via the gut axis.
Pirfenidone: Pirfenidone - Wikipedia