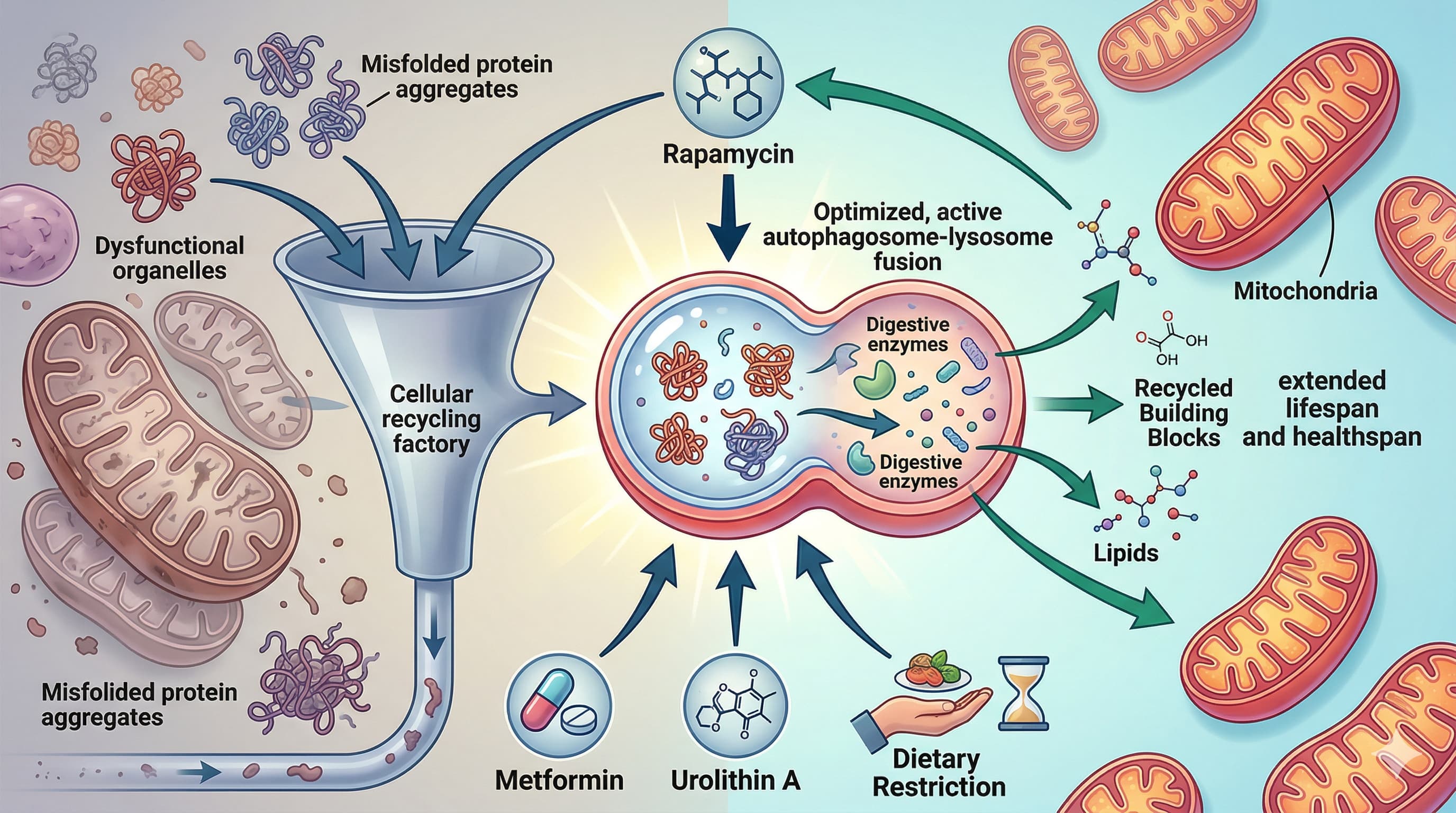
The ultimate holy grail in longevity research isn’t a silver bullet; it is the optimization of a cellular trash disposal system known as autophagy. A 2026 review explicitly links the lifespan-extending benefits of popular pharmacological agents (like rapamycin, metformin, and urolithin A) and lifestyle interventions (dietary restriction, exercise, sleep, and temperature stress) directly to this conserved cellular recycling mechanism. What makes this paradigm-shifting is the mounting evidence that these seemingly disparate biohacking strategies are just different physiological roads leading to the exact same metabolic destination: clearing out misfolded proteins and dysfunctional organelles to prevent age-related decline.
While the causative role of autophagy in lifespan extension is irrefutable in model organisms like yeast, nematodes, and mice—where knocking out autophagy genes completely erases the benefits of fasting or rapamycin—human data remains stubbornly correlational. The core roadblock is the field’s current inability to accurately measure dynamic “autophagic flux” in living human tissues, forcing researchers to rely on noisy, indirect proxies like peripheral blood mononuclear cell (PBMC) profiling.
Despite this analytical gap, the paper highlights actionable insights for human healthspan. From early time-restricted feeding that syncs autophagy to circadian rhythms, to the mechanical stress of resistance training triggering Chaperone-Assisted Selective Autophagy (CASA) in muscle tissue, we are learning precisely how to activate this pathway in an organ-specific manner. Even temperature stress is gaining mechanical clarity, with cold exposure driving systemic lipophagy via hypothalamic signaling. Ultimately, stacking these interventions could provide synergistic healthspan benefits, even as we wait for the clinical flux assays needed to quantify them perfectly in living humans.
Institution: This research originates from The Buck Institute for Research on Aging in the USA,
Journal: published in the Journal of Molecular Biology.
*Impact Evaluation: The impact score of this journal is 4.7, evaluated against a typical high-end range of 0–60+ for top general science, therefore this is a Medium impact journal.
Type: Comprehensive Literature Review evaluating In vivo, In vitro, and Clinical trial data.
Subjects: Cross-species analysis aggregating data from Saccharomyces cerevisiae (yeast), Caenorhabditis elegans(nematodes), Drosophila melanogaster (fruit flies), Mus musculus (mice), Rattus norvegicus (rats), and human clinical cohorts.
Lifespan Analysis
Lifespan Data: As a review covering decades of research, absolute lifespan extension metrics vary across the aggregated studies. However, the paper establishes that lifespan extensions achieved by rapamycin, spermidine, and dietary restriction are unequivocally negated when core autophagy genes (e.g., Atg5, Atg7, BECN1) are genetically silenced.
Mechanistic Deep Dive
The authors outline how disparate interventions converge on specific cellular nodes:
mTORC1 & AMPK Axis: Caloric restriction and rapamycin directly inhibit mTORC1, a potent negative regulator of autophagy, allowing the ULK1 initiation complex to trigger phagophore formation. Conversely, metformin and endurance exercise activate AMPK, which directly phosphorylates ULK1 while simultaneously inhibiting mTORC1.
Chaperone-Assisted Selective Autophagy (CASA): Resistance training exerts mechanical stress on muscle fibers, upregulating a specialized, mechanosensitive form of autophagy (CASA) that recycles force-bearing cytoskeletal proteins. This process operates independently of standard endurance-triggered macroautophagy, strongly suggesting that concurrent training (cardio + heavy resistance) offers additive proteostatic benefits across different muscle fiber types [Confidence: High].
Systemic Exerkines: Acute endurance exercise induces the secretion of Fibronectin 1 (FN1) from skeletal muscle into the bloodstream. This circulating exerkine acts as an inter-tissue communication molecule that forces autophagy activation in distant organs, primarily the liver.
Circadian Integration: Dietary restriction strategies, such as intermittent time-restricted feeding (iTRF), fail to extend lifespan if circadian clock genes or core autophagy genes are knocked out, proving that nutrient timing is as mechanistically critical as nutrient deprivation.
Temperature-Driven Lipophagy: Cold exposure triggers preliminary neuronal signaling in the hypothalamus that communicates with brown adipose tissue (BAT) via the peripheral nervous system, initiating lipophagy (autophagic breakdown of lipid droplets) to fuel thermogenesis.
Novelty
This 2026 review aggregates fragmented biohacking paradigms into a unified autophagic theory of aging. It moves beyond the classic “starvation equals autophagy” model to mechanistically map how chronic sleep fragmentation dysregulates amyloid clearance in the brain via enlarged, dysfunctional lysosomes. Furthermore, it identifies how temperature stress elicits highly tissue-specific selective autophagic cascades (e.g., BAT lipophagy) rather than just a generalized, whole-body cellular stress response.
Critical Limitations
Human Translational Gap: The paper bluntly acknowledges the glaring lack of a reliable, non-invasive assay to measure autophagic flux in living humans. Static measurements of LC3-II protein levels in human muscle biopsies or PBMC blood draws are easily confounded; a buildup of autophagosomes can indicate either robust autophagy activation or a pathological bottleneck in lysosomal degradation [Confidence: High].
Over-activation Toxicity: The biological assumption that “more autophagy is always better” is heavily flawed. The review notes that prolonged or extreme dietary restriction overactivates starvation signals, leading to autophagy-dependent organismal death.
Non-Canonical Pathway Ambiguity: The field has heavily focused on classic macroautophagy, largely ignoring alternative ATG5/ATG7-independent pathways or non-canonical functions of ATG8 (like CASM, where ATG8 conjugates to single membranes), which are poorly understood but clearly linked to inflammation and neurodegeneration. It remains entirely unknown how human lifestyle factors modulate these alternative pathways [Confidence: Medium].
Age-Dependent Efficacy: Older human subjects exhibit blunted autophagic responses to both heat and cold stress compared to younger cohorts. It remains unproven whether an older individual derives the same molecular benefit from a cold plunge as a young adult, and this age-related impairment may severely limit the therapeutic utility of temperature stress later in life [Confidence: High].
Really interesting to see the detail of this paper. In terms of specific actionable advice I asked Grok to summarize and it generated:
Here’s a clear summary of the specific advice and findings on lifestyle factors that influence autophagy, drawn directly from the review “Links Between Autophagy and Healthy Aging” by Ebata and Hansen (2026). The paper emphasizes that these factors often induce autophagy, which helps mediate benefits for healthspan and lifespan in model organisms (worms, flies, mice), with some supportive (but more correlative) evidence in humans. Autophagy is frequently shown to be causal via genetic knockdown experiments.
1. Dietary Restriction / Balanced Diet
Caloric restriction (CR) or dietary restriction (DR): Moderate reduction in calorie intake (e.g., ~30% CR in humans for 3–15 years) increases expression of autophagy genes such as BECN1 and LC3B/ATG8 in skeletal muscle. In model organisms, DR strongly induces autophagy (more autophagosomes, higher Atg8/LC3 markers) and requires autophagy genes (e.g., Atg5, Atg7, bec-1/BECN1, atg-7) for lifespan extension. It works partly via mTOR inhibition and AMPK/TFEB activation. Practical note: Moderate DR reduces oxidative stress and inflammation; excessive restriction can be harmful (over-activation of autophagy may lead to negative outcomes in some models).
Intermittent fasting (IF) or time-restricted feeding (TRF): Especially early-day TRF (eTRF) or night-biased iTRF in flies optimizes circadian-timed autophagy and improves metabolic health/glucose regulation. In humans, eTRF enhances autophagy markers, lowers inflammation, and improves glucose levels.
CR mimetics like spermidine: Found in foods such as wheat germ, soybeans, and nuts. It induces autophagy (via eIF5A hypusination and TFEB) and extends lifespan in yeast, flies, worms, and mice while suppressing age-related autophagy decline in human immune cells (T/B cells). It also reduces senescent cells.
Overall advice: A balanced diet with periods of reduced nutrient intake (CR/IF) or spermidine-rich foods promotes autophagy. Combine with other factors for synergy; avoid extremes.
2. Exercise
Endurance exercise (e.g., swimming in worms, running/swimming in mice): Increases autophagosomes, LC3-II/LC3-I ratio, and reduces p62 in muscle, heart, and brain. Requires autophagy/mitophagy genes (unc-51/ULK1, bec-1/BECN1, pink-1, etc.) for lifespan and healthspan benefits. Chronic voluntary exercise prevents age-related decline in autophagy markers (LC3, ATG7, BECN1) especially in type II muscle fibers. Systemic effects occur via muscle-secreted factors like FN1 (fibronectin 1).
Resistance exercise: Induces chaperone-assisted selective autophagy (CASA) via BAG3/HSC70/HSPB8 in human skeletal muscle. Progressive loading appears more effective than constant load.
Human evidence: Acute or chronic exercise alters plasma autophagy markers (e.g., BECN2, LC3B-II). Serum from exercised mice induces autophagy in human cells.
Practical advice: Regular endurance and resistance training counteracts the age-related drop in autophagy, improves mitochondrial function, and reduces inflammation. Benefits are seen across ages but may be stronger when started earlier or maintained consistently.
3. Sleep Adjustments
Sufficient, high-quality, and regular sleep: Maintains proper autophagy dynamics and circadian alignment. In model organisms, sleep deprivation or fragmentation impairs brain autophagy (accumulated autophagosomes, reduced flux to autolysosomes, enlarged lysosomes), leading to cognitive decline, neurodegeneration (e.g., amyloid-β buildup), and proteostasis failure.
Disruption harms autophagy: Chronic sleep fragmentation disrupts circadian autophagy rhythms and mimics aged profiles. Knockdown of autophagy genes (Atg1/ULK1, Atg8) can alter sleep duration, suggesting bidirectional regulation. Chaperone-mediated autophagy (CMA) loss also disrupts circadian rhythms.
Human data: Limited and mixed — short-term severe restriction (4 hours/night) showed little change in muscle autophagy markers in small studies.
Practical advice: Prioritize consistent sleep schedules and sufficient duration/quality to support autophagy and brain proteostasis. Align with circadian rhythms (e.g., via timed eating + sleep) for better metabolic and longevity outcomes. Disrupted sleep accelerates brain aging via impaired clearance.
4. Temperature Modulation (Hormetic Stress)
Mild heat stress: Short hormetic heat (e.g., 36°C in worms) extends lifespan and requires autophagy genes (unc-51/ULK1, bec-1, lgg-1). Increases autophagy flux. In humans, warm bathing (40°C) or sauna use (80–100°C) may boost autophagy markers (e.g., p62 changes) and is linked to better cardiovascular health and lower mortality risk.
Cold exposure: Moderate cold (e.g., 4°C in mice or mild core temperature drop in humans) induces autophagy (higher LC3B-II) in brown fat, liver, and immune cells, including lipophagy for thermogenesis. Requires autophagy genes (Atg5, Becn1). Benefits are hormetic but often impaired in older individuals (lower response in ~60s vs. ~20s men).
Practical advice: Incorporate safe hormetic temperature challenges — such as sauna sessions, warm baths, or cold showers/baths — to stimulate autophagy. Effects may be stronger in younger people or when combined with other interventions; older adults might need gentler approaches.
Key Takeaways and Caveats from the Paper
These lifestyle factors commonly induce or preserve autophagy, which is a shared mechanism behind many healthy-aging benefits (reduced inflammation, better mitochondrial function, proteostasis).
Evidence is strongest (causal) in model organisms via autophagy gene requirements; in humans it is mostly correlative due to challenges in measuring autophagy flux non-invasively.
Best results: Combine interventions (e.g., time-restricted eating + exercise) for potential synergy.
Gaps: More reliable human biomarkers and longitudinal studies are needed. Individual differences (age, sex) matter — e.g., older adults may show blunted responses to cold.
These recommendations align with a general healthy lifestyle: balanced eating patterns (with occasional restriction), regular physical activity, good sleep hygiene, and occasional mild temperature stresses. Always consult a healthcare professional before making significant changes, especially with fasting or extreme temperatures.
Yes - you are correct that the paper identifies many compounds and activities that stimulate autophagy… BUT… given that it seems that when you knockout the autophagy genes, the lifespan benefit goes away… it is then obvious that because rapamycin is by far and away the best life extension molecule, it must also have by far the best autophagy profile. So it seems that there are quickly diminishing returns after rapamycin, in terms of autophagy activators.
Yes the paper does give a lot of support to rapamycin as the most reproducible, and widest acting/broadest autophagy inducer. And puts the focus of autophagy for its core benefits. And
supports its benefit over other autophagy strategies as aging blunts the body’s autophagic systems.
But I think it still leaves lots of really interesting questions:
Are alternative strategies additive is a key one. Or does an optimal dose of rapamycin optimize autophagy?
Urolithin A and Spermidine use very different pathways and serve very different mechanistic functions - so for someone with the genes to benefit from them - it may be better to combine. It’s just we have no way of knowing.
We’re obviously shooting in the dark here but my fortnightly rapamycin may be too infrequent - or it may be optimal (or too much) because I’m also: time restricted eating, taking berberine, urolithin a (pom peel) and spermidine (wheat germ), sauna-ing and exercising.
And things may change as I age and my autophagy from these other things becomes “blunted” by age.
Given the massive uncertainty, I’m disinclined to put all my eggs in one autophagic basket - probably for no good reason - but i still prefer my current scattergun approach. Urolithin A/Spermidine (through Pomegranate Peel amd wheat germ in my daily yoghurt) is likely safe, likely beneficial even whilst I’m on fortnightly rapamycin. And it “feels” less risky than weekly rapamycin.
If I were older, this paper might encourage me to be a bit bolder with my rapamycin though.