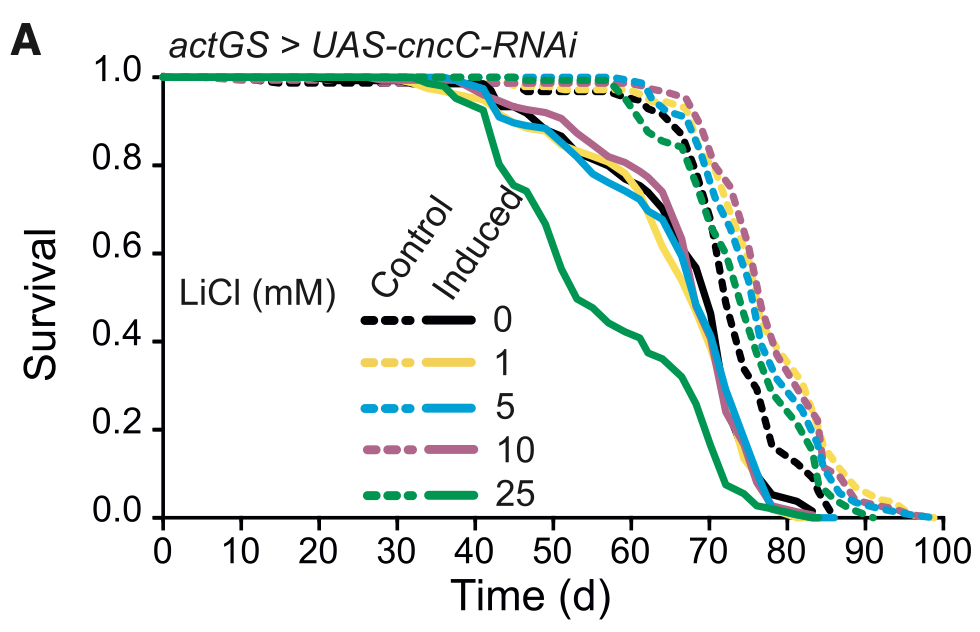
With a handful of people on this site reporting that they are taking lithium for longevity (and also quite a few people on Reddit taking it for biohacking or longevity purposes), it’s something I’ve started looking into myself.
I wanted to call attention to some papers supporting a novel longevity mechanism of lithium: genomic stability. The first two papers show directly an effect of lithium on genomic stability, although they don’t provide evidence that this occurs through the lithium target GSK3β. The next three papers show that GSK3β inhibition may increase genomic stability, and although they don’t use lithium, they provide indirect evidence that lithium will do the same. In the third part of this post I’ll try to provide some evidence that subclinical lithium doses can effectively inhibit GSK3β.
Onto the papers…
- Direct Effects of Lithium on Genomic Stability
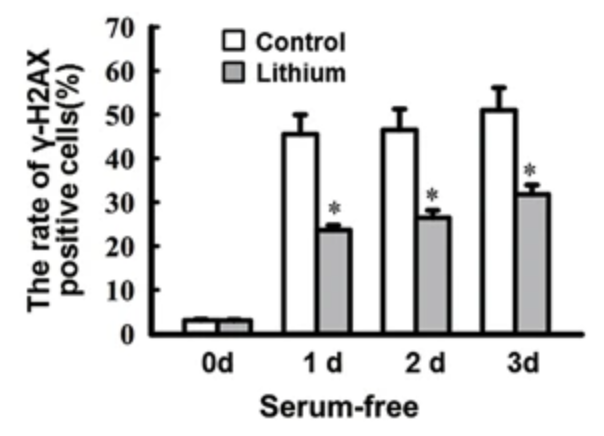
In primary rat retinal neurocytes, serum deprivation led to an increased number of phosphorylated H2AX-positive cells, while pretreatment with 1mM LiCl (0.16mM lithium) reduced the number of number of γH2AX-positive cells. As γH2AX is generally considered to represent unrepaired double-strand breaks/DSBs, this data suggests that lithium increases the efficiency of DSB repair.
The primary pathway of DSB repair is nonhomologous end-joining/NHEJ, and in the serum deprived cells, lithium gave a 3x efficiency increase in the NHEJ assay.
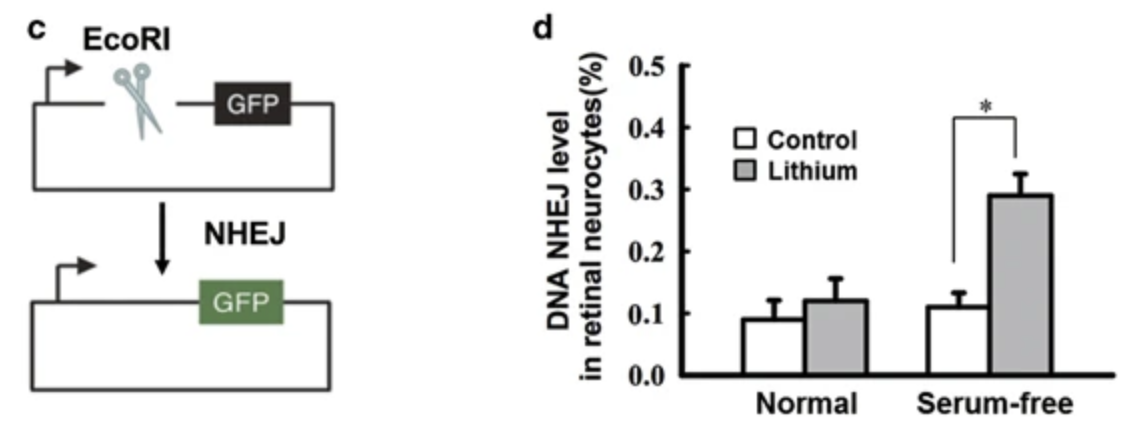
Lithium led to a 3x increase in LIG4 protein levels (this is a DNA ligase which helps to reconnect the separated DNA strands in the NHEJ pathway) and this was proposed as the mechanism leading to increased DNA stability.
A second study showed that a week of pretreatment with 3mM lithium in HT-22 cells (mouse hippocampal cell line) could reduce comet tail formation in response to radiation, which is a powerful inducer of DSBs. These comet tails are formed in response to DNA damage, with the ‘mean tail moment’ reflecting the degree of damage. In this same cell line, lithium enhanced repair efficiency in the NHEJ assay, and they found this NHEJ enhancement to require phosphorylation of Thr2609 on DNA-PK.
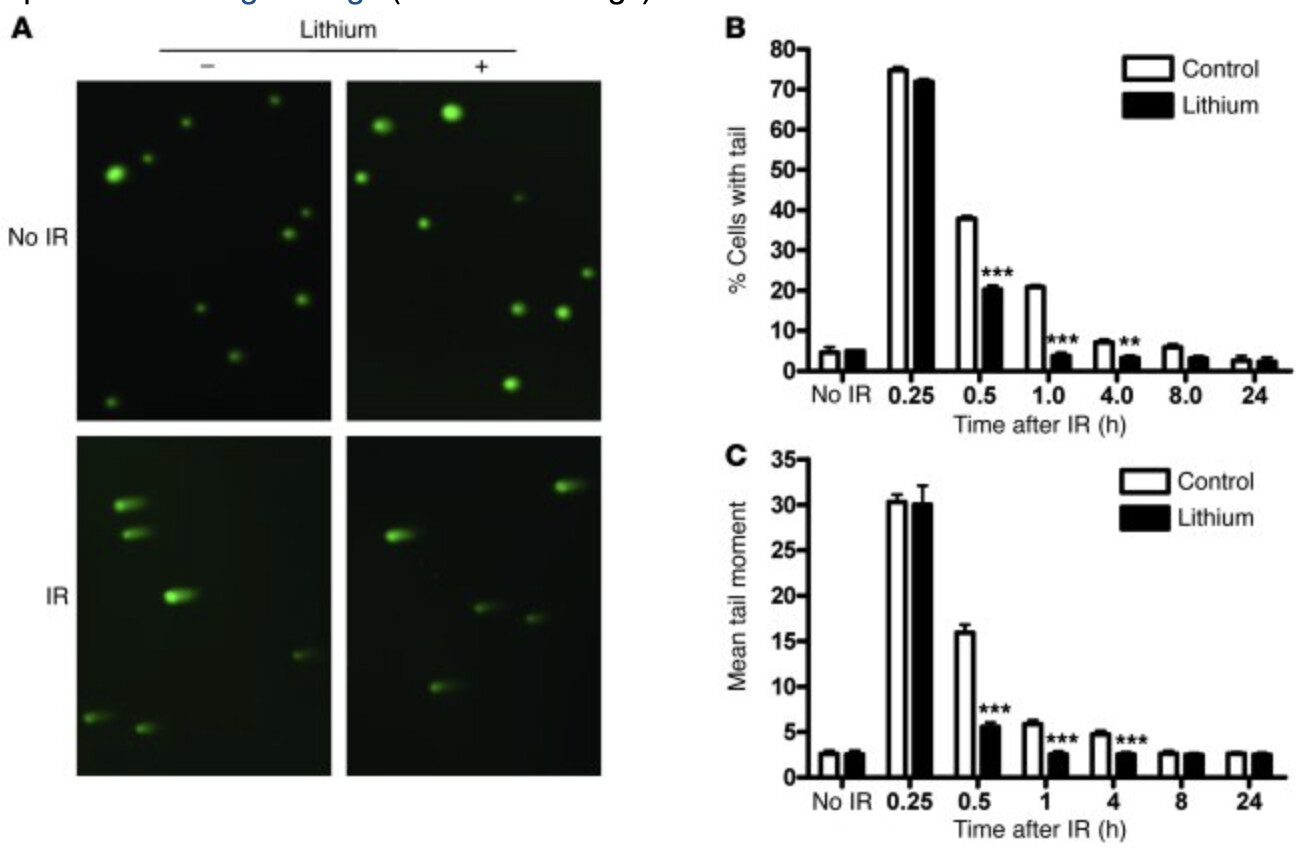
They then showed that similar lithium pretreatment reduced the persistence of γH2AX foci, in either HT-22 cells or primary neurons exposed to radiation. Additionally, in the primary neurons, lithium reduced the number of γH2AX foci per cell. Similar activity was demonstrated in-vivo, as 40mg/kg ip for 7 days reduced the number of γH2AX-positive hippocampal cells in irradiated mice. This study found no effect of lithium on homologous recombination, an important DSB repair pathway in dividing cells.
- Direct Role of GSK3β in Genomic Stability
We present evidence to suggest that the stimulation of A2AR markedly facilitated DNA repair through the TRAX/DISC1/GSK3β complex in a rat neuronal cell line (PC12), primary mouse neurons, and human medium spiny neurons derived from iPSCs. A2AR stimulation led to the inhibition of GSK3β, thus dissociating the TRAX/DISC1/GSK3β complex and facilitating the non-homologous end-joining pathway (NHEJ) by enhancing the activation of a DNA-dependent protein kinase via phosphorylation at Thr2609. Similarly, pharmacological inhibition of GSK3β by SB216763 also facilitated the TRAX-mediated repair of oxidative DNA damage
Specifically, this study showed that inhibition of GSK3β accelerated double strand-break (DSB) repair efficiency in irradiated mouse hippocampal neurons, as assessed by the neutral comet assay. This coincided with attenuation of IR-induced γ-H2AX foci, a well characterized in situ marker of DSBs. To confirm the effect of GSK3 activity on the efficacy of DSB repair, we further demonstrated that biochemical or genetic inhibition of GSK3 activity resulted in enhanced capacity in nonhomologous end-joining–mediated repair of DSBs in hippocampal neurons. Importantly, none of these effects were observed in malignant glioma cells
Here, we showed that TRAX participates in the ATM/H2AX-mediated DNA repair machinery by interacting with ATM and stabilizing the MRN complex at double-strand breaks.
- Potency of Lithium’s GSK3β inhibition/Intracellular Lithium Concentrations Achieved with sub-mM Serum Levels
One of lithium’s primary targets is GSK3β, where lithium’s Ki is 2mM. It’s also worth knowing that GSK3β is constitutively active, so its activity is regulated by inhibitory phosphorylations and/or trapping within protein complexes.
As lithium appears to inhibit GSK3β via competition with the magnesium co-factor binding site, it’s been argued that lithium’s in-vivo GSK3β IC50 is 0.8mM or less.
Let’s say someone taking daily lithium for longevity has serum levels of 0.05mM, is this a dose that will actually inhibit GSK3β? We can look to a study in rats, which showed that with chronic dosing, lithium concentrates intracellularly. After 3 weeks of daily administration, intracellular levels in the brain were over 4x higher than CSF or serum levels. [ref]
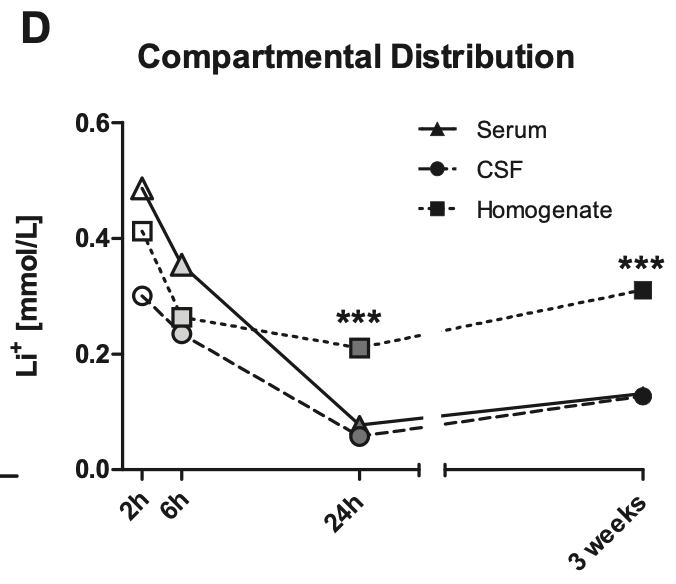
Let’s assume then that with chronic dosing, a serum level of 0.05mM corresponds to concentration of 0.2mM within brain cells. This would correspond to 20% inhibition of GSK3β, assuming a GSK3β IC50 of 0.8mM. [ref]
The actual level of GSK3β inhibition produced by 0.2mM lithium might be greater than 20% though, see for example
The lithium-induced increase in serine9-phosphorylation of GSK3β may be critical because it provides a mechanism whereby a moderate direct inhibitory effect of lithium achieved by therapeutically relevant lithium concentrations is amplified to achieve more pronounced inhibition of GSK3β by the therapeutic level of lithium [ref]
Moreover, the act of serum deprivation in the above study rapidly leads to loss of GSK3β-ser9 phosphorylation, and if you recall the first paper at the top of this post (lithium both reduces γH2AX-positive cells and enhances NHEJ assay efficiency in serum deprived media), this further supports the idea that lithium activates DNA repair pathways via GSK3β.