Actually I don’t know precisely what it was.
1 Like
Interesting observation: CIRBP (the protein responsible for the bowhead whale’s extremely efficient and accurate DSB repair) is actually a substrate for GSK3β.
While CIRBP normally localizes to the nucleus, phosphorylation by GSK3β and CK2 partially redistributes it to the cytosol. Off topic: CK2 is also an interesting kinase, and may mediate the monoamine release of amphetamines by favoring channel-like monoamine transporter conformations.
As GSK3β is normally cytosolic, it’s unclear what effect (if any) lithium would have on CIRBP activity. But regardless, I’m going to take any opportunity I can to mention CIRBP, as it’s a potential home-run longevity target.
The bowhead whale is among the longest-lived mammals (maximum lifespan north of 200 years), and its extremely efficient and accurate CIRBP-dependent DSB repair (apparently due to strong CIRBP expression relative to other mammals) possibly contributes significantly to this.
While accurate DSB repair will prevent loss of protein-coding sequences and non-coding RNA sequences, as well as their regulatory sequences, an efficient and rapid repair of DSBs would also be expected to dampen the loss of epigenetic information. This can be seen with the Sinclair lab’s ICE system, which showed in vivo that non-mutagenic DSBs still lead to changes in gene expression and DNA methylation, and that these changes resemble those seen in aging.
Essentially, the act of repairing DSBs seems to requires and/or is associated with chromatin states which are permissive with respect to epigenetic drift. The faster we can fix them, the less time we spend in these states of accelerated epigenetic aging.
My hand-wavy analogy for this would be a potential well. The youthful chromatin states are analogous to the local energy minimum, and they can’t readily transition to aged chromatin states (the global energy minimum) because of the energy barrier. The DSB repair response, by altering the interactions between DNA and DNA-binding proteins, lowers this energy barrier and favors transition to aged chromatin states.

3 Likes
Although I accept that DSB repair is important, I don’t see how DSB repair failure would create the well known and predictable aging phenotype. I can see it causing a general gradual breakdown of transcription, but not a targeted breakdown.
1 Like
I personally find the relocalization of chromatin modifiers (RCM) hypothesis is one of the most convincing mechanisms of epigenetic aging (and also aging in general), and there’s evidence that DSBs are involved, in a manner that seems mostly independent of their ability to produce insertions and deletions.
The exact nature of DSB-induced changes in chromatin structure and how these structural changes might impart age-associated transcriptional changes (accelerated elongation and increased stalling of RNA polymerase) isn’t clear, but it’s plausible that changes in chromatin structure could produce such a transcriptional phenotype, or partially contribute to it.
Despite this, the primary contributor to age-associated RNA polymerase stalling appears to be DNA damage, perhaps of an oxidative variety.
Subsequently, we assessed whether various potential parameters were correlated with the degree of GLPT to identify a mechanism explaining RNAPII stalling. We did not find significant differences between GLPThigh genes and other gene categories (transcriptionally upregulated and downregulated; remainder) in nucleotide content across gene bodies, transcriptional error rate, alternative splicing, chromatin accessibility, histone modifications associated with euchromatin or DNA methylation patterns, which would point to epigenetic changes being responsible (Extended Data Figs. 4–6). These factors do not correlate with the degree of age-related GLPT, which is expected when such a factor is causally involved, and hence do not explain the observed transcriptional decline.
In view of gene-length-dependent transcriptional stalling, a plausible explanation is accumulation of transcription-blocking DNA damage because long genes have a higher probability to acquire stochastic lesions26,29. Therefore, we monitored de novo RNA synthesis in the livers of Xpg −/− mice, which display many features of widespread premature aging and a 20-week life span due to defects in the DNA repair pathways TCR and global genome nucleotide excision repair by which they are unable to remove transcription-stalling lesions34. Both EU staining and EU-seq at the age of 7 and 14 weeks (Fig. 4a–c) revealed an age-dependent, progressive, pan-nuclear decline in transcription. EU-seq in premature aging global genome nucleotide excision repair and TCR-defective Ercc1 Δ/− mice that display more severe liver aging pathology due to an additional defect in interstrand cross-link repair35,36, exhibited already high transcription loss at 4 and even more at 10 weeks (Fig. 4d), showing that the extent of transcriptional decline, DNA repair deficiency and severity of liver pathology are correlated. [ref]
As stated above, Xpg is a component of the nucleotide excision repair pathway, and is known to remove oxidative lesions like 5-guanidinohydantoin and spiroiminodihydantoin (both of which form from 8-oxoguanine), and both of which have been shown to stall RNA polymerase II.
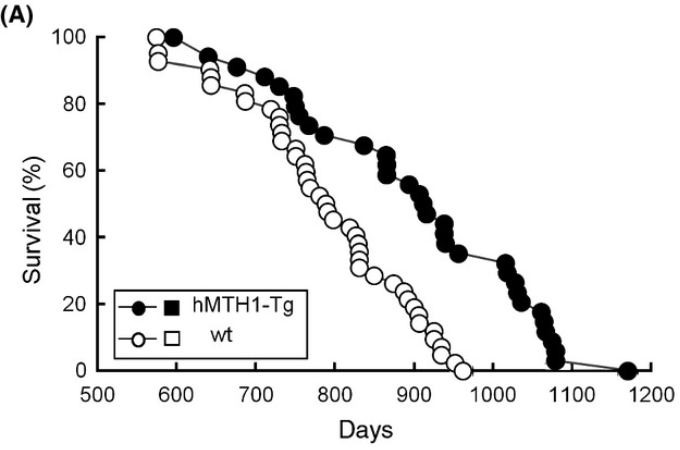
This is supported by increased lifespan in hMTH1 transgenic mice, although ideally this would be shown in genetically heterogenous mice with control median lifespan >850 days
hMTH1-Tg mice express high levels of the hMTH1 hydrolase that degrades 8-oxodGTP and 8-oxoGTP and excludes 8-oxoguanine from both DNA and RNA.
…
hMTH1-Tg animals live longer. Their median lifespan was 4 months greater than that of their wild-type littermates (914 vs. 790 days) (P < 0.0004; Kaplan Meier test) (Fig. 1A). [ref]
Also interesting to note that the age-associated changes in elongation rate appear to arise from histone and nucleosome depletion. As DNA G-quadruplexes have been shown to increase with replicative aging and form on nucleosome-free DNA, I wonder if they might be contribute to RNAPII stalling.
There’s not a ton of research on this and G-quadruplexes close to the transcription start site appear to stimulate transcription. OTOH, they can also produce an absolute transcriptional block, and I wonder if G-quadruplexes further downstream in the gene body might produce stalling. Presumably the average number of G-quadruplexes in the gene body would be proportional to its length, so this could explain the length-dependence of transcriptional changes.
4 Likes
I think the stalling relates in some way to a wait for substrate possibly also linked to methylation of the DNA. this will happen more frequently in long genes. However, i need to read your references.
2 Likes
Indeed it may be that changes in elongation rate arise from histone/nucleosome depletion. I think here it is best to consider what the evolutionary merit of the histone is.
As I see it having the histone and the ability to open up and close down DNA has one primary advantage in that it protects the DNA from harm by wandering molecules. If there were to be no histone then there would be considerably more DNA damage.
When DNA is transcribed ordinarily it is opened up via the process of acetylation then transcribed and sealed up again. I have a hypothesis that the reason why germline DNA suffers less damage is that it is transcribed less frequently and in metabolically less active cells. Hence the chance of it having damage is lower rather than a better repair system.
Now if you lose the nucleosomes then the DNA becomes open, that is open to both transcription and damage. It may be possibly that the G-quadruplexes act as a back up system to protect the DNA, but not as effectively as the histone. At the same time you would expect transcription to speed up as the need for acetylation disappears.
This then raises the question as to whether histone depletion is a secondary effect of aging or a primary cause.
In essence, however, I don’t think epigenetic information is “lost” per se, but that through metabolic problems the genome is not functioning at its optimal level. Hence if you improve the metabolism (through SOX2 one of the Yamanaka factors or some other mechanism) the genome starts functioning properly again.
The histone itself may be primarily generated in the S phase, but there is an interesting question as to whether a form of histone homeostasis exists in well functioning cells which means that histone depletion is a consequence of that system breaking down.
It strikes me as likely that there would be some system for maintaining the histone. However, I have only done a cursory search.
In the end, however, if we are looking at the failure to transcribe long genes. There is the splicing factors theory, there is the DNA damage theory (which perhaps links to the papers cited) and my own hypothesis is that it arises from a metabolic cause.
In many ways I think the fact that adjusting the metabolism fixes at least part of the problem at a macroscopic level gives strength to the metabolic argument.
However, it is a worthwhile discussion to be had.
1 Like
Separately I thought I would find a reference to the RCM
1 Like
The histone itself may be primarily generated in the S phase, but there is an interesting question as to whether a form of histone homeostasis exists in well functioning cells which means that histone depletion is a consequence of that system breaking down.
I wouldn’t be surprised if histone depletion is tied to impaired nuclear import of histones, due to dysfunctional nuclear pore complexes/NPCs.
It’s pretty well-accepted that impaired nucleocytoplasmic compartmentalization (NCC) occurs in cellular aging, and as minimal NPC biogenesis occurs in post-mitotic cells, they might be especially susceptible to impaired NCC. Some of the NPC components (e.g Nup93) have extremely long lifetimes, and might be needed to last the entire lifetime of the cell. This implies that it’s unlikely that post-mitotic cells could recover from damage to these components, at least under physiological conditions.
There’s a number of small-molecule rejuvenating cocktails that have been shown to reverse these changes in NCC. Would be interesting to use fluorescent tags to see whether nuclear to cytoplasmic ratio of histone H3 and H4 decreases with aging (i.e whether it’s an issue with nuclear import) and if so, which cocktails reverse these changes.
1 Like
Do you think citrate transport inhibition effect would bring possibly specific positive or negative effects?
Fabio
Citrate transport inhibition would create a negative effect. I think this is the mechanism that causes renal damage at high lithium concentrations.
1 Like

For people taking lithium, has anyone looked into the safety profile of lithium for long-term use? It seems there are some papers out there emphasizing kidney damage from lithium, even at 1 to 5mg low doses.
1 Like
FWIW: N=1. I have been taking lithium orotate for ~30 years (average 10 mg elemental lithium daily), and my kidney functions are very good for any age and excellent for 85.
There are always Chicken Little skeptics for any supplement we might be taking.
I don’t think that lithium orotate is dangerous long-term for doses under 20 mg daily. The preponderance of papers I have looked at has convinced me that lithium orotate supplementation is a good thing.
2 Likes
Also depends on what else you’re taking. For example, SGLT2i flush lithium out, while telmisartan accumulates it. You can’t just look at a drug/supplement/diet in isolation, you must see it in the total context. In one context it could be helpful, but in another harmful. For most people 1-5 mg lithium orotate will not be toxic.
2 Likes
If there are papers please link to two of them.
My personal anecdote: I have been taking anywhere between 1-10mg lithium for 5-6 years and my Cystatin C (gold standard kidney health blood test) is at the very bottom of the range at 0.64 (0.60-1.0), which is exactly what one should want.
3 Likes