Actionable Intelligence
The Translational Protocol (Rigorous Extrapolation)
-
Human Equivalent Dose (HED):
-
Calculation: Using the FDA Guidance for Estimating the Maximum Safe Starting Dose, the translation from Cynomolgus monkey to adult human (assuming 60 kg) utilizes Body Surface Area (BSA) normalization factors (Km). Monkey Km = 12; Human Km = 37.
-
Math: 30 mg/kg * (12 / 37) = 9.73 mg/kg.
-
Human Dose: For a standard 70 kg adult, the target HED is roughly 680 mg/day. This falls well within standard supplemental ranges and avoids the necessity of intravenous administration.
-
Pharmacokinetics (PK/PD):
-
Bioavailability: Vitamin C exhibits dose-dependent, active transport kinetics via SVCT1 and SVCT2 receptors. At an oral dose of 500–1000 mg, bioavailability is approximately 70% to 80%, but it steeply declines at higher doses due to tissue saturation and renal clearance. Peak plasma concentrations plateau at approximately 70–80 micromolar.
-
Half-life: Highly variable depending on baseline plasma levels, typically ranging from 10 to 20 hours. Reference: NIH ODS.
-
Safety & Toxicity:
-
LD50: The oral LD50 in rats is 11,900 mg/kg.
-
NOAEL / Upper Limit: The established Tolerable Upper Intake Level (UL) in humans is 2,000 mg/day. Toxicity at this level is generally limited to osmotic diarrhea and gastrointestinal disturbances.
-
Phase I Safety Profile: Vitamin C is generally recognized as safe (GRAS). However, high-dose continuous supplementation increases urinary oxalate excretion, elevating the risk of calcium oxalate kidney stones in susceptible individuals.
-
CYP450 / Liver / Kidney: No significant CYP450 inhibition or induction. Primary clearance is renal. Extreme doses in patients with pre-existing renal impairment (or G6PD deficiency) can induce hemolysis or acute oxalate nephropathy.
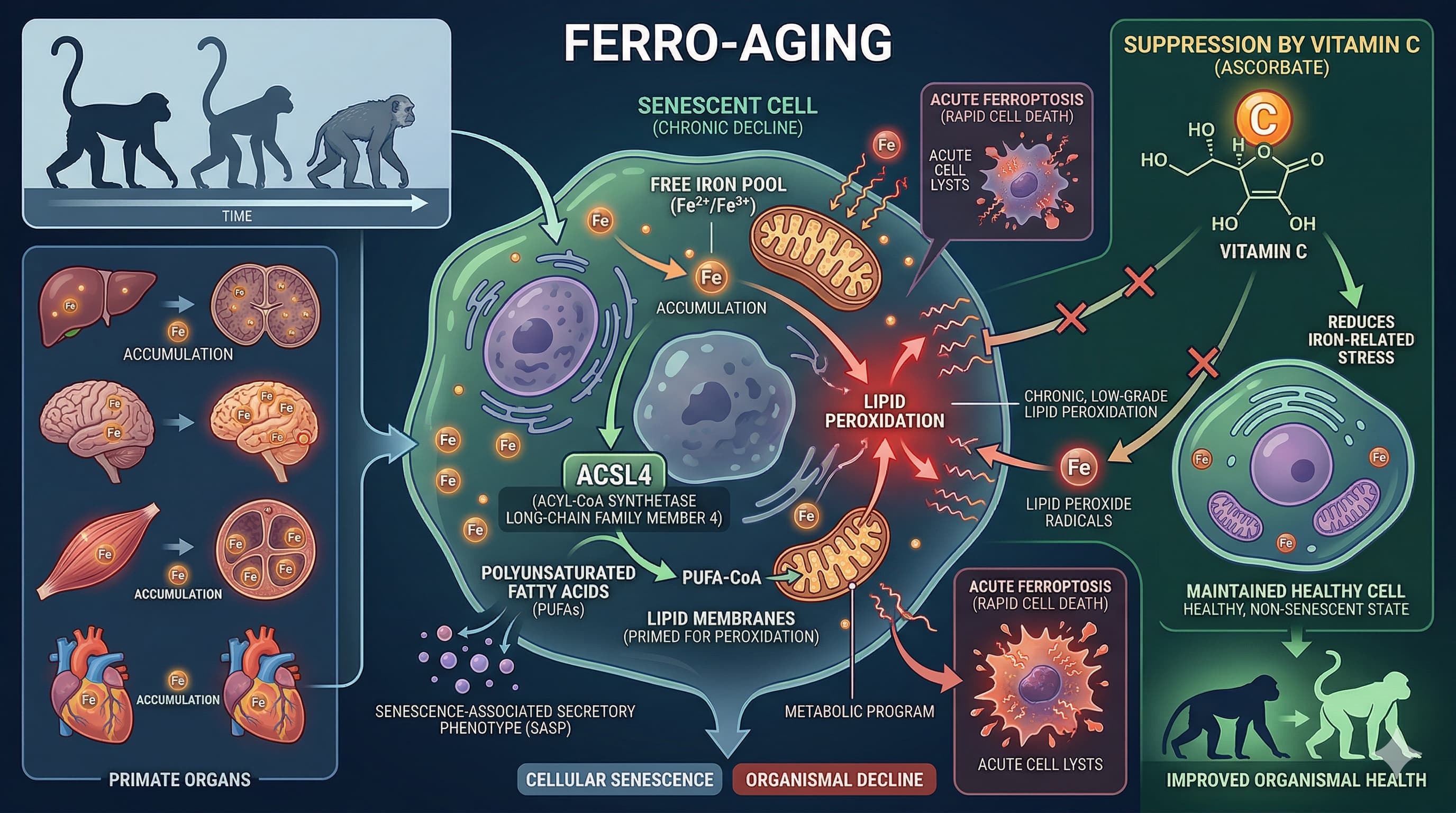
Biomarker Verification To verify target engagement of ACSL4 inhibition in a clinical or biohacking setting, track the following downstream lipidomic and oxidative markers in plasma or PBMCs (Peripheral Blood Mononuclear Cells):
-
Direct ACSL4 Metabolites: Reduced levels of Arachidonoyl-CoA (20:4-CoA) and Adrenoyl-CoA (22:4-CoA).
-
Downstream Phospholipids: Decreased polyunsaturated phosphatidylcholines (PC) and phosphatidylethanolamines (PE), specifically PE 38:4 and PE 36:4.
-
Lipid Peroxidation End-Products: Reduced systemic malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE).
Feasibility & ROI
-
Sourcing: Over-the-counter (OTC). Ubiquitously available as ascorbic acid.
-
Cost vs. Effect: Monthly cost for a high-quality 500-1000 mg/day supplement is under $10. Given the minimal cost and high safety profile, the ROI is massive if the ACSL4 inhibition translates clinically to humans. It is a highly practical addition to a longevity protocol.
Part 5: The Strategic FAQ
1. If Vitamin C is a potent ACSL4 inhibitor, why have previous large-scale, high-dose longevity and cardiovascular RCTs failed to show significant lifespan extension? Answer: Previous trials viewed Vitamin C purely as a systemic antioxidant rather than a targeted enzyme inhibitor. It is highly probable that typical supplemental dosing achieves plasma saturation (approx. 80 micromolar) but fails to reach the intracellular concentrations required to competitively inhibit the ACSL4 binding pocket across all tissue types, particularly in organs with high lipid turnover. Furthermore, generic antioxidant trials often fail because they indiscriminately suppress necessary hormetic ROS signaling.
2. Does Vitamin C have a sufficient binding affinity (Kd) to outcompete endogenous polyunsaturated fatty acids for the ACSL4 active site at physiological concentrations? Answer: The paper’s computational docking and in vitro assays suggest successful competition, but in vivo enzymatic kinetics are missing. Since humans actively regulate plasma Vitamin C, achieving the necessary intracellular stoichiometry to outcompete abundant arachidonic acid in lipid-rich environments (like the brain) without intravenous administration remains a critical knowledge gap.
3. The macaques received 30 mg/kg/day, but their baseline diet already contained roughly 150 mg/day. Did this study just cure captive primate scurvy/sub-clinical deficiency? Answer: Captive primates require exogenous Vitamin C, just like humans. While the authors claim the control group was not deficient, doubling the baseline intake may simply optimize a baseline physiological deficit rather than inducing a novel, supra-physiological geroprotective state. We need data comparing this dose to a known, strictly optimized baseline cohort.
4. ACSL4 is essential for normal membrane remodeling and cellular function. Does chronic, long-term inhibition risk membrane instability or impaired cellular repair? Answer: Yes. Complete knockout of ACSL4 is often embryonically lethal or highly deleterious in pre-clinical models. The goal is partial attenuation, not total ablation. Chronic, high-level inhibition could theoretically impair necessary inflammatory responses, macrophage activation, and membrane repair processes.
5. How does Vitamin C interact with Rapamycin, considering both modulate lipid metabolism and cellular senescence? Answer: Rapamycin inhibits mTOR, triggering autophagy and altering lipid profiles (often causing transient hyperlipidemia). ACSL4 inhibition by Vitamin C targets the downstream consequence of lipid membrane oxidation. There is no acute pharmacological conflict, and they likely act synergistically by targeting distinct hallmarks of aging (nutrient sensing vs. oxidative macromolecular damage).
6. Metformin relies on mild complex I inhibition and transient ROS generation to trigger AMPK. Will high-dose Vitamin C blunt this effect? Answer: This is a known translational risk. High-dose antioxidants (specifically Vitamins C and E) have been shown in multiple human trials to blunt the insulin-sensitizing and mitochondrial-biogenesis effects of exercise and potentially metformin by quenching the necessary ROS signal. Timing doses away from exercise and metformin administration is highly recommended. Reference: Antioxidants prevent health-promoting effects of physical exercise in humans (2009).
7. Does this ACSL4 inhibition mechanism interfere with SGLT2 inhibitors or Acarbose? Answer: No direct pharmacokinetic or pharmacodynamic conflicts exist. SGLT2 inhibitors (e.g., empagliflozin) act hemodynamically and metabolically via the kidneys, while acarbose acts in the gut. Vitamin C’s intracellular action on ACSL4 should run parallel without interference.
8. Can Vitamin C cross the blood-brain barrier in sufficient quantities to reverse the neuro-degeneration (brain atrophy) observed in the primate models? Answer: Vitamin C crosses the blood-brain barrier via the SVCT2 transporter in its reduced form, and via GLUT1 in its oxidized form (dehydroascorbic acid), where it is then reduced back. The brain maintains some of the highest concentrations of Vitamin C in the body, making targeted ACSL4 inhibition in neural tissue theoretically feasible with oral dosing.
9. The study highlights reduced cGAS-STING activation. Is this a direct effect of Vitamin C or downstream of ACSL4 inhibition? Answer: It is downstream. ACSL4 drives lipid peroxidation, which damages mitochondrial and nuclear membranes, leading to cytosolic DNA leakage. The cGAS-STING pathway senses this misplaced DNA and triggers sterile inflammation. By halting the upstream membrane damage, Vitamin C indirectly silences the cGAS-STING inflammaging alarm.
10. What specific human data is required to move this from a “promising primate study” to a “verified human longevity protocol”? Answer: We need a placebo-controlled RCT in middle-aged to older adults tracking the specific lipidomic biomarkers identified above (20:4-CoA, PE 38:4), alongside epigenetic clock analysis, over a 12-to-24-month period. Until we see targeted ACSL4 metabolite reduction in human plasma, this remains an informed hypothesis.