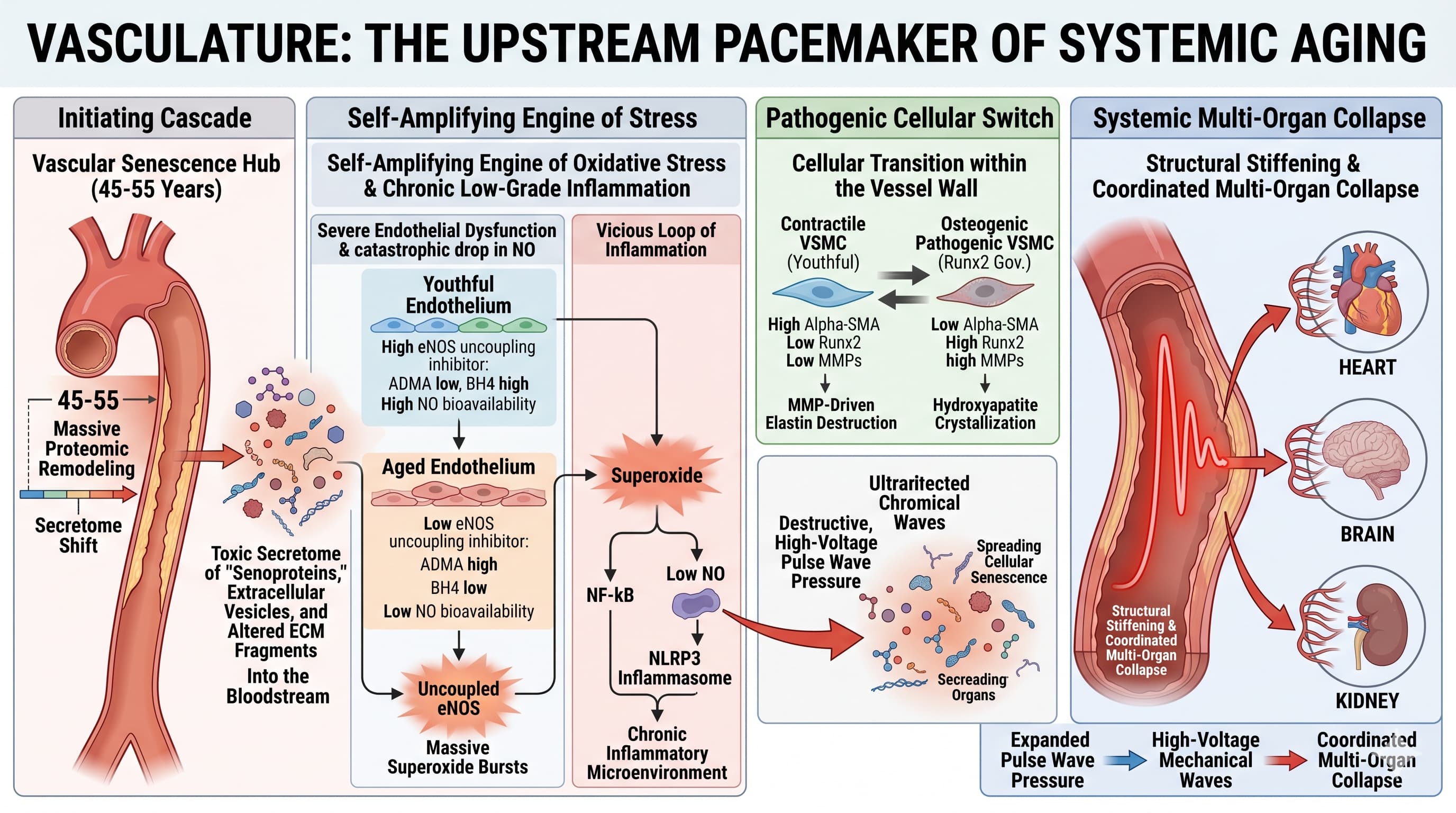
Potentially Actionable Interventions Ideas & Evidence
Some ideas to investigate that may help mitigate the issues outlined in the paper.
1. MitoQ (Mitoquinol Mesylate)
The Core Strategy
MitoQ is a mitochondria-targeted antioxidant comprising a ubiquinone moiety covalently bound to a lipophilic triphenylphosphonium (TPP+) cation. This structural modification utilizes the highly negative mitochondrial membrane potential to drive a 100-fold to 1000-fold accumulation of the compound directly inside the inner mitochondrial membrane. MitoQ specifically targets the mitochondrial reactive oxygen species (mtROS) burst that drives age-related endothelial nitric oxide synthase (eNOS) uncoupling. By continuously scavenging mitochondrial superoxide anions, MitoQ preserves tetrahydrobiopterin (BH4) from oxidation, restores homeostatic eNOS homodimer coupling, and maximizes endothelial nitric oxide (NO) bioavailability. The intended longevity outcome is the suppression of upstream endothelial “inflammaging,” preservation of microvascular perfusion, and a long-term reduction in central arterial stiffness.
Translational Dosing Protocol
Preclinical rodent models typically administer MitoQ via drinking water at concentrations up to 250 micromolar, approximating a dose of 3.5 mg/kg/day in mice. Human clinical protocols have bypassed purely theoretical math due to direct validation in human clinical trials. However, back-calculating from the mouse model using Body Surface Area (BSA) normalization demonstrates close translational alignment:
-
Formula: HED = Animal Dose * (Animal Km / Human Km)
-
Raw Math: HED = 3.5 mg/kg/day * (3 / 37) = 3.5 * 0.0811 = 0.284 mg/kg/day
-
Human Dose Calculation: 0.284 mg/kg/day * 70 kg = 19.88 mg/day
This aligns precisely with the clinically validated human dose of 20 mg orally once daily, taken in the morning on an empty stomach.
-
Pharmacokinetics: Oral bioavailability is highly limited but enhanced by the lipophilic TPP+ homing construct. Peak plasma concentrations (Cmax) are achieved within 1 hour post-ingestion. The systemic elimination half-life is approximately 12 to 15 hours.
Literature Validation & Source Verification
In a double-blind, randomized, placebo-controlled crossover clinical trial evaluating healthy older adults aged 60 to 79 years, 6 weeks of oral MitoQ supplementation at 20 mg/day successfully reversed microvascular endothelial dysfunction. MitoQ treatment drove a 42% increase in brachial artery flow-mediated dilation (FMD)—a standard clinical proxy for vascular tissue integrity—which is equivalent to an approximate 7-year reduction in vascular biological age. This rescue was mediated by a 25% reduction in plasma oxidized low-density lipoprotein (oxLDL) levels and a corresponding drop in systemic oxidative stress biomarkers. Review the data registries directly via the Hypertension Study on Mitochondrial Antioxidant Supplementation in Older Adults and the Chronic Mitochondrial Antioxidant Treatment Protocol via American Journal of Physiology.
Safety, Toxicity, & Interaction Profile
-
Safety Metrics: The No Observed Adverse Effect Level (NOAEL) established in chronic rodent toxicological models is 20 mg/kg/day. In human trials, mild gastrointestinal distress (nausea, abdominal discomfort) was documented in fewer than 10% of participants at the therapeutic 20 mg/day threshold.
-
Organ Toxicity: No adverse hepatic transaminase elevation or acute kidney injury signals (elevated creatinine or blood urea nitrogen) have been documented at standard clinical dosing.
-
CYP450 Interactions: MitoQ does not significantly inhibit or induce major Cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2D6, or CYP3A4) at therapeutic systemic exposures.
Longevity Stack Compatibility
-
Rapamycin: Fully compatible; complements rapalog therapy by buffering localized mitochondrial oxidative stress without interfering with the downstream anti-proliferative signaling of mTORC1.
-
SGLT2 Inhibitors / Metformin: Highly compatible; displays synergistic endothelial protection via concurrent mitochondrial ROS suppression and AMPK-driven metabolic resetting.
-
Acarbose / 17-alpha Estradiol: No known structural or functional contraindications.
-
PDE5 Inhibitors (Tadalafil/Sildenafil): Synergistic; by increasing raw NO bioavailability, MitoQ enhances the upstream substrate pool for cyclic guanosine monophosphate (cGMP), potentially magnifying the vasodilatory and endothelial compliance outputs of PDE5 inhibitors.
2. Ultra-Low-Dose Terazosin (YTHDF2 Inhibitor / ATF3 Stabilizer)
The Core Strategy
Terazosin is historically classified as an alpha-1 adrenergic receptor antagonist utilized for benign prostatic hyperplasia and hypertension. However, recent epitranscriptomic discovery isolates a non-canonical, blood pressure-independent utility: terazosin binds to and inhibits the N6-methyladenosine (m6A) RNA reader protein YTHDF2. In the aging vasculature, hyperactivation of the methyltransferase METTL14 drives excessive m6A methylation and subsequent YTHDF2-mediated degradation of Activating Transcription Factor 3 (Atf3) mRNA inside vascular smooth muscle cells (VSMCs). By blocking YTHDF2, terazosin stabilizes Atf3 mRNA transcripts. ATF3 directly binds to the Atg7 promoter to upregulate Autophagy-Related 7 (Atg7) transcription, activating a protective ATF3-ATG7 autophagy loop. This non-hemodynamic mechanism restores autophagic clearance of cellular debris, halts the phenotypic switch of VSMCs from a contractile to a pathogenic osteogenic state, and suppresses smooth muscle cell senescence.
Translational Dosing Protocol
To achieve targeted YTHDF2 inhibition and Atf3 mRNA stabilization without triggering alpha-1 receptor-mediated systemic hypotension in normotensive individuals, the clinical protocol demands an ultra-low, sub-therapeutic dose of 0.5 mg to 1.0 mg orally once daily, strictly administered at bedtime. Back-calculating from the protective dose utilized in mouse models of senescence (approximately 0.15 mg/kg/day):
-
Formula: HED = Animal Dose * (Animal Km / Human Km)
-
Raw Math: HED = 0.15 mg/kg/day * (3 / 37) = 0.15 * 0.0811 = 0.0122 mg/kg/day
-
Human Dose Calculation: 0.0122 mg/kg/day * 70 kg = 0.854 mg/day
This positions a 1.0 mg/day tablet as the ideal translatable target dose.
-
Pharmacokinetics: Oral absorption is rapid with near 90% bioavailability. Food does not alter absolute exposure. Peak plasma concentrations occur within 1 hour, and the systemic elimination half-life is 12 hours.
Literature Validation & Source Verification
Multi-omics validation pipelines and single-cell RNA sequencing models have established that terazosin treatment interrupts the METTL14-YTHDF2 degenerative cascade. In aged mouse models, low-dose terazosin stabilized Atf3 transcript pools, enhanced absolute autophagic flux, significantly reduced lipid-rich plaque areas, and reversed carotid-femoral pulse wave velocity (cfPWV) independent of systemic blood pressure modifications. Review the direct mechanistic evidence in the Advanced Science Study on ATF3 and VSMC Senescence and the European Heart Journal Discovery on METTL14 Inhibition and Vascular Aging.
Safety, Toxicity, & Interaction Profile
-
Safety Metrics: The rodent oral NOAEL is 5 mg/kg/day. In humans, the primary side effect of terazosin is orthostatic hypotension, which is strictly dose-dependent and highly minimized at the 0.5 mg to 1.0 mg bedtime threshold.
-
Organ Toxicity: No structural hepatic or renal toxicity signals are evident in long-term human databases. Terazosin undergoes extensive hepatic metabolism via dimerization and oxidation, with roughly 40% eliminated via biliary excretion in feces and 60% excreted in urine.
-
CYP450 Interactions: Terazosin is not a substrate, inhibitor, or inducer of major Cytochrome P450 isoforms, reducing the risk of competitive enzymatic clearance.
Longevity Stack Compatibility
-
Rapamycin / Metformin: Fully compatible; works in parallel with rapamycin-driven mTORC1 inhibition and metformin-driven AMPK activation to maximize distinct nodes of the systemic autophagy network.
-
SGLT2 Inhibitors: Safe; however, because SGLT2i molecules induce mild osmotic diuresis and transient blood pressure reduction, concurrent ultra-low-dose terazosin use requires close monitoring of orthostatic blood pressure stability.
-
PDE5 Inhibitors (Tadalafil/Sildenafil): Critical Contraindication / High Risk. Co-administration of alpha-blockers with PDE5 inhibitors can trigger severe, additive, peripheral vasodilation culminating in acute orthostatic hypotension and syncope. If tadalafil is included in the longevity stack, terazosin must be withheld, or separated by a minimum 24-hour pharmacokinetic window with frequent hemodynamic tracking.
3. High-Dose Vitamin K2 (Menaquinone-7 / MK-7)
The Core Strategy
Vitamin K2 as Menaquinone-7 (MK-7) serves as an obligatory cofactor for the gamma-glutamyl carboxylase enzyme. In the vascular microenvironment, its primary target is the post-translational activation of Matrix Gla Protein (MGP)—a potent local inhibitor of ectopic medial vascular calcification. Uncarboxylated MGP (dp-ucMGP) is biologically inert, allowing unchecked calcium phosphate crystal deposition along elastic fibers and promoting the transdifferentiation of smooth muscle cells into osteoblasts via Runx2 upregulation. Fully carboxylated MGP directly binds and sequesters calcium crystals, stabilizes the arterial extracellular matrix (ECM), and inhibits bone morphogenetic protein-2 (BMP-2) osteogenic signaling. The intended longevity outcome is the cessation and potential reversal of arterial media calcification, thereby improving large artery compliance and lowering pulse wave velocity.
Translational Dosing Protocol
While standard nutritional requirements target 90 to 120 mcg/day for basic coagulation homeostasis, vascular-targeted calcification inhibition demands a high-dose therapeutic protocol of 360 mcg orally once daily, consumed alongside a fat-containing meal to maximize lipophilic lymphatic uptake. Preclinical rodent intervention architectures deploy 100 micrograms per gram of MK-7 per diet weight, generating massive system exposures that map onto high human clinical requirements.
-
Pharmacokinetics: MK-7 possesses near 100% absorption metrics. Due to its long hydrocarbon side chain, it incorporates into low-density lipoproteins, achieving an extended plasma elimination half-life of approximately 50 to 72 hours (compared to a brief 1 to 2 hour half-life for Vitamin K1). This enables sustained extrahepatic tissue distribution and uninterrupted vascular MGP engagement.
Literature Validation & Source Verification
In longitudinal human clinical screenings, long-term high-dose MK-7 supplementation significantly altered large-artery mechanical compliance. A 3-year double-blind randomized controlled trial utilizing healthy postmenopausal women demonstrated that a daily oral intake of MK-7 stopped the progression of age-related arterial stiffening. In individuals exhibiting high baseline vascular stiffness, high-dose MK-7 therapy drove a statistically significant regression of arterial stiffness, measured via clinical carotid-femoral pulse wave velocity (cfPWV) and reflection indices. Review the complete clinical protocols and data registries in the VitaK-CAC Trial on Menaquinone-7 and Vascular Calcification and the Danish InterVitaminK Population-Based Protocol via PMC.
Safety, Toxicity, & Interaction Profile
-
Safety Metrics: Vitamin K2 as MK-7 exhibits no established upper tolerable intake level or No Observed Adverse Effect Level (NOAEL) constraints; human and animal models demonstrate a lack of systemic toxicity even at multi-milligram doses. MK-7 does not alter baseline coagulation profiles or cause a hypercoagulable state in individuals not taking vitamin K antagonists, as the carboxylation of hepatic clotting factors saturates at low nutritional intakes.
-
Organ Toxicity: No renal or hepatic adverse events have been reported.
-
CYP450 Interactions: MK-7 does not demonstrate induction or competitive inhibition across standard human hepatic CYP450 enzyme pools.
Longevity Stack Compatibility
-
Rapamycin / SGLT2 Inhibitors / Metformin / Acarbose / 17-alpha Estradiol: Fully compatible; no known direct or indirect negative interactions.
-
Warfarin (Coumadin): Absolute Contraindication. High-dose MK-7 will directly outcompete and neutralize the vitamin K antagonist mechanism of warfarin, precipitating a drop in international normalized ratio (INR) levels and creating a high thromboembolic risk. This contraindication does not apply to Direct Oral Anticoagulants (DOACs like apixaban or rivaroxaban).
4. Empagliflozin (SGLT2 Inhibitor)
The Core Strategy
Empagliflozin selectively inhibits the Sodium-Glucose Cotransporter 2 (SGLT2) within the proximal renal tubules, lowering systemic glucose levels through glucosuria. However, its core vascular longevity value operates through non-glycemic metabolic and anti-inflammatory pathways. Empagliflozin triggers a systematic “fasting mimicry” state that downregulates nutrient-sensing growth networks and upregulates the AMP-activated protein kinase (AMPK) pathway. Activated AMPK directly stimulates endothelial cell autophagy, forcing the clear-out of senescent mitochondrial networks, reducing intracellular lipid droplet retention, and maintaining the structural integrity of the endothelial glycocalyx. Downstream, this suppressive profile inhibits the assembly of the NLRP3 inflammasome, directly halting the maturation and endocrine release of the pro-atherogenic senoproteins IL-1beta, IL-18, and IL-6. The intended longevity outcome is the optimization of endothelial function, stabilization of vulnerable atherosclerotic plaques, and prevention of heart failure with preserved ejection fraction (HFpEF).
Translational Dosing Protocol
To achieve robust endothelial AMPK activation and suppress vascular inflammaging, the protocol utilizes standard clinical dosing: 10 mg orally once daily, taken in the morning with or without food.
-
Formula (Theoretical Back-Calculation): HED = Animal Dose * (Animal Km / Human Km)
-
Raw Math: HED = 10 mg/kg/day * (6 / 37) = 10 * 0.1622 = 1.622 mg/kg/day
-
Human Dose Calculation: 1.622 mg/kg/day * 70 kg = 113.5 mg/day
Note: Because empagliflozin acts as an uncompetitive active transport inhibitor in the human kidney with vastly higher affinity for human SGLT2 than rodent configurations, human metabolic saturation occurs completely at 10 mg/day, which overrides the theoretical animal surface area math.
-
Pharmacokinetics: Empagliflozin is rapidly absorbed following oral administration, displaying an absolute bioavailability of approximately 85%. Peak plasma levels are achieved at 1.5 hours post-dose. It exhibits a prolonged terminal elimination half-life of approximately 12.4 hours. Metabolic clearance is driven by glucuronidation via 5’-adenosine diphosphate-glucuronosyltransferases (UGT2B7, UGT1A9, and UGT1A1); it bypasses hepatic Cytochrome P450 systems.
Literature Validation & Source Verification
Large-scale cardiovascular outcome trials (EMPA-REG OUTCOME) have confirmed that empagliflozin reduces major adverse cardiovascular events (MACE) and cardiovascular mortality by 38%. Mechanistic animal studies reveal that empagliflozin preserves endothelial function, reduces absolute vascular wall permeability, and maintains plaque stability in ApoE-/- models on a high-fat diet purely via AMPK-mediated autophagy upregulation. Review the source material in the Cardiovascular Research Study on SGLT2i and Plaque Stability and the Journal of Physiology and Biochemistry Analysis on Empagliflozin-Induced Autophagy.
Safety, Toxicity, & Interaction Profile
-
Safety Metrics: The established preclinical oral NOAEL is 100 mg/kg/day in rats. In human cohorts, the primary safety concern is a 3-fold to 4-fold increase in genitourinary tract infections (vulvovaginal candidiasis, balanitis) secondary to high urinary glucose concentrations. A rare but critical risk is euglycemic diabetic ketoacidosis (eDKA), particularly during periods of acute infection, prolonged fasting, or perioperative windows.
-
Organ Toxicity: Empagliflozin reduces glomerular hyperfiltration, displaying a long-term nephroprotective signature that slows the progression of chronic kidney disease (CKD).
-
CYP450 Interactions: Bypasses the CYP450 cascade; no major interactions are reported with standard CYP inhibitors or inducers.
Longevity Stack Compatibility
-
Rapamycin: High Synergistic Compatibility. Chronic rapalog use can induce off-target metabolic dysregulation, including hyperglycemia and hyperlipidemia. Empagliflozin counteracts rapamycin-induced insulin resistance, maintains glycemic control, and limits rapalog-induced lipid accumulation without disrupting the structural inhibition of mTORC1.
-
Metformin: Synergistic; couples with metformin to drive robust, dual-node activation of the AMPK complex, amplifying systemic metabolic flexibility.
-
Acarbose / 17-alpha Estradiol / PDE5 Inhibitors: Fully compatible; no negative chemical or physiological cross-talk documented.
Part 2: Strategic Feasibility & Target Engagement
Biomarker Verification
To verify that these vascular interventions are successfully engaging their targets and delaying systemic senescence, individuals must track a multi-dimensional array of functional, structural, and circulating biomarkers:
-
Functional Metrics: Target engagement of the endothelial NO pathway is verified by performing office-based brachial artery Flow-Mediated Dilation (FMD), targeting an absolute dilation score greater than 5%. Large artery compliance and matrix remodeling are monitored via Carotid-Femoral Pulse Wave Velocity (cfPWV), looking for an absolute reduction in measured centimeters per second over a 6-to-12 month tracking arc.
-
Structural Biomarkers: A non-contrast cardiac CT scan provides the Coronary Artery Calcification (CAC) Agatston Score. Target engagement of Vitamin K2 therapy is verified by a deceleration or arrest of the annual CAC score progression velocity below the standard 15% rapid-advancement threshold.
-
Circulating SASP Factors: Systemic vascular inflammaging is monitored by tracking plasma High-Sensitivity Interleukin-6 (hs-IL-6) and Growth Differentiation Factor 15 (GDF-15) levels. Successful intervention must demonstrate a progressive downward trend in these core senoprotein markers.
-
Translational Target Engagement Assays: Vitamin K2 matrix engagement is quantified via enzyme-linked immunosorbent assays measuring plasma dephosphorylated-uncarboxylated MGP (dp-ucMGP); a sharp drop in circulating dp-ucMGP indicates successful peripheral tissue gamma-carboxylation. Endothelial cell senescence can be monitored via liquid biopsy tracking of Endothelial Extracellular Vesicles (EVs) expressing surface protein arrays for VE-cadherin and p21.
Sourcing & Financial ROI
The table below outlines the procurement tier, estimated monthly expenditure, direct product linking, and a comparative cost-to-benefit ratio for maintaining the calculated longevity protocol:
| Intervention |
Sourcing Status |
Estimated Monthly Cost (USD) |
Product Page URL |
Cost-to-Benefit Ratio & Clinical Justification |
MitoQ
(20 mg/day)
|
Dietary Supplement
(Over-the-Counter)
|
$104.99 |
Welltopia Pharmacy MitoQ 20mg |
High ROI. Readily accessible acellular compound backed by direct human clinical trial data demonstrating reversal of microvascular endothelial aging in older adults. |
Terazosin
(1.0 mg/day)
|
Prescription (Rx Only)
(Generic / Off-Label)
|
$6.99 – $9.00 |
GoodRx Terazosin Price Coupon |
Exceptional ROI. Extremely inexpensive generic alternative. Offers a targeted mechanism for stabilizing the ATF3-ATG7 autophagy feedback loop to clear out senescent smooth muscle cell populations at a nominal cost. |
Vitamin K2 (MK-7)
(360 mcg/day)
|
Dietary Supplement
(Over-the-Counter)
|
$0.60 – $1.20 |
BulkSupplements Vitamin K2 MK7 |
High ROI. Fills a unique therapeutic niche as a validated inhibitor of active arterial medial calcification. Utilizing raw bulk powder maximizes financial economy while delivering therapeutic-grade dosing. |
Empagliflozin
(10 mg/day)
|
Prescription (Rx Only)
(Brand / Off-Label)
|
$354.86 |
GoodRx Jardiance Price Coupon |
Moderate-to-High ROI. Represents a significant financial expense if paid out-of-pocket without a Type 2 Diabetes diagnosis. However, the clinical value is exceptional for individuals over age 50, providing upstream multi-organ preservation, systemic AMPK activation, and protection against rapalog-induced metabolic drift. |
Shipping Cost Summary
-
MitoQ (Welltopia Pharmacy): Free standard shipping is automatically applied within the United States for orders over $50.00, meaning this intervention incurs $0.00 in additional shipping fees.
-
Vitamin K2 MK7 (BulkSupplements): Standard domestic flat-rate shipping is $5.95 for orders under $59.00, and free standard shipping is unlocked on all orders totaling $59.00 or more. Purchasing a multi-month supply at once completely eliminates the shipping premium.
-
Terazosin & Empagliflozin (GoodRx / Local Retail Pickup): Because these are prescription-only medications obtained via GoodRx coupons at physical retail pharmacies (e.g., Walgreens, CVS, CostCo), there are zero shipping costs involved. For optional mail-order home delivery through integrated platforms like HealthWarehouse, standard ground shipping is fully included at a $0.00 flat rate for prescription items.