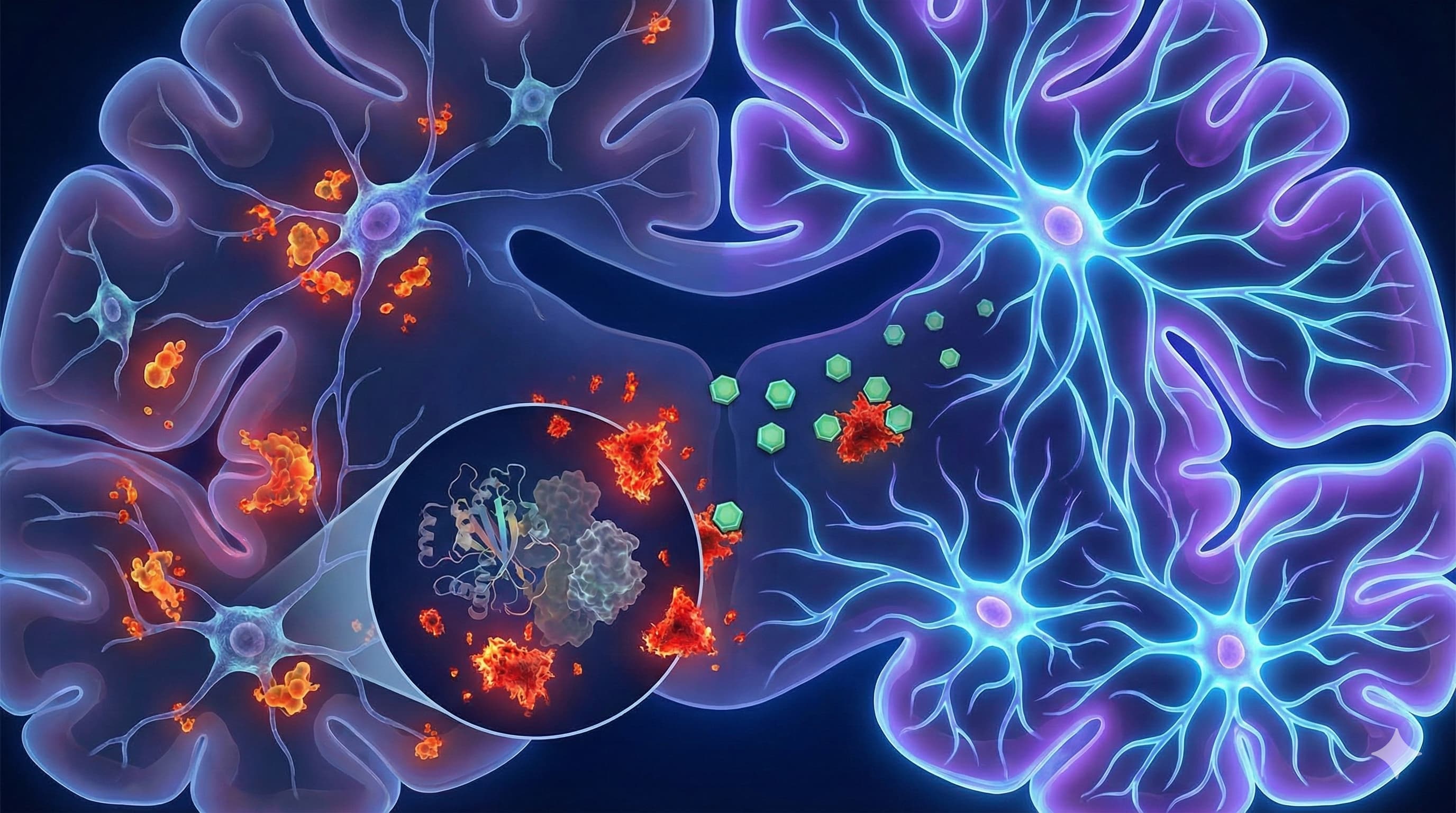
In a comprehensive 2026 review published in Cells, Dr. Masaru Tanaka presents a strategic “roadmap” that shifts the longevity paradigm from merely suppressing inflammation to actively reprogramming the dialogue between the immune system and the brain. The “Big Idea” here is that age-related cognitive decline isn’t just about neurons dying; it’s about the “niche” environment—specifically microglia—stopping the conversation that tells stem cells to build new neurons. Tanaka argues that microglia don’t just become “inflamed” with age; they lose their specific “pro-neurogenic” programming, effectively shutting down the brain’s repair shop.
The paper identifies five critical “gaps” that currently block the translation of longevity science into human therapy: the lack of longitudinal data (tracking the same brain over time), the undefined heterogeneity of glial cells (not all microglia are the same), the persistence of “innate immune memory” (bad habits stuck in the cell’s DNA), the ignored vascular component, and the poor alignment between mouse models and human biology.
Tanaka proposes a “playbook” to bridge these gaps, highlighting NLRP3 inflammasome inhibitors and epigenetic editing as the most promising near-term interventions. He argues that by selectively inhibiting the NLRP3 pathway (which drives the “sterile inflammation” of aging), we can break the maladaptive feedback loops without compromising the immune system’s ability to fight infection—a crucial distinction for life-extension strategies.
Source:
- Open Access Paper: Neurogenesis and Neuroinflammation in Dialogue: Mapping Gaps, Modulating Microglia, Rewiring Aging
- Context: Neuroscience Research Group, Hungarian Research Network, University of Szeged, Hungary / MDPI, Switzerland. Journal: Cells.
- Impact Evaluation: The impact score of this journal is 5.2 (2024 JIF), evaluated against a typical high-end range of 0–60+ for top general science, therefore this is a Medium-High impact specialty journal.
Part 2: The Biohacker Analysis
Study Design Specifications:
- Type: Review & Theoretical Perspective (Not a primary in vivo study).
- Subjects: Synthesizes data from Murine (Mouse/Rat) models and Human clinical pathology.
- Lifespan Data: Cites external data (e.g., progeroid mice lifespan extension with NLRP3 inhibitors) but presents no new lifespan curves of its own.
Mechanistic Deep Dive:
-
The Core Shift: The paper details the transition of the “Neurogenic Niche” from a Supportive State (Young) to a Toxic State (Old).
- Young: Microglia secrete BDNF, IGF-1, and IL-4, actively stimulating Neural Stem Cells (NSCs) to proliferate.
- Old: “Inflammaging” causes a switch. Microglia pump out IL-1β and TNFα. Crucially, IL-1β suppresses neurogenesis directly by locking NSCs in a quiescent (dormant) state or forcing them to become astrocytes (scar tissue) instead of neurons.
- The Pathway: The NLRP3 Inflammasome is identified as the central engine of this toxicity. It senses “danger signals” (cellular debris, mitochondrial DNA) accumulating in the aging brain and triggers the release of IL-1β.
- Organ Priority: Hippocampus (Dentate Gyrus)—the seat of memory and spatial navigation, and one of the few places humans grow new neurons.
Novelty: What distinguishes this paper is the “5 Gaps” Framework. Instead of just saying “inflammation is bad,” Tanaka systematically categorizes why anti-inflammatory trials fail (e.g., lack of spatial resolution, cross-species mismatch) and proposes Glial Reprogramming—using CRISPR or specific inhibitors to “reset” microglia to their youthful, BDNF-secreting state rather than just killing them or shutting them off.
Critical Limitations:
- Review Status: This is a synthesis of other people’s work. It generates hypotheses, not data.
- The “Microglia” Conundrum: The paper admits that distinguishing “resident microglia” from “infiltrating macrophages” is technically difficult in humans. Treatments targeting one might inadvertently suppress the other, impairing peripheral immunity.
- Translational Gap: While it hypes NLRP3 inhibitors, it relies heavily on mouse data where high doses (200mg/kg) are used. The specific “reprogramming” protocols (CRISPR for microglia) are years away from clinical viability.
Part 3: Actionable Intelligence
Actionable Intelligence (Deep Retrieval & Validation Mode) Instruction: Extrapolating the paper’s focus on NLRP3 inhibition to the leading clinical candidate, Dapansutrile (OLT1177).
The Translational Protocol (Rigorous Extrapolation):
- Compound: Dapansutrile (OLT1177) (Oral, selective NLRP3 inhibitor). Note: Do NOT use MCC950 (hepatotoxic).
-
Human Equivalent Dose (HED):
- Mouse Data: Effective neuroprotection/lifespan extension seen at ~200 mg/kg (i.p.) or dietary equivalent (~3.75 g/kg diet).
- Calculation: 200 mg/kg (mouse) × (3/37) ≈ 16.2 mg/kg (human).
- For 70kg Human: ~1,135 mg/day.
- Clinical Trial Reality: Phase 2 trials for acute gout use 1,000 mg BID (2,000 mg/day) or a loading dose of 2,000 mg.
- Longevity Protocol (Speculative): Lower chronic dosing (e.g., 300–500 mg/day) is hypothesized by biohackers but untested for safety.
-
Pharmacokinetics:
- Bioavailability: High (>90% in humans).
- Half-life: ~22–26 hours (Supports once-daily dosing).
- Brain Penetration: Yes, crosses the Blood-Brain Barrier (Crucial for neurogenesis).
Safety & Toxicity Check:
- Safety Profile: OLT1177 is considered safe in Phase 1/2 trials, unlike its predecessor MCC950.
-
Toxicity Signals:
- Liver: No significant hepatotoxicity observed in trials (major advantage over MCC950).
- Immunity: Potential increased risk of infection (it dampens the innate immune response). Monitor for signs of upper respiratory infection.
- Contraindications: Active infections (bacterial/viral).