Digging more into the specific medications and supplements mentioned in the paper:
Hard stops discovered during research:
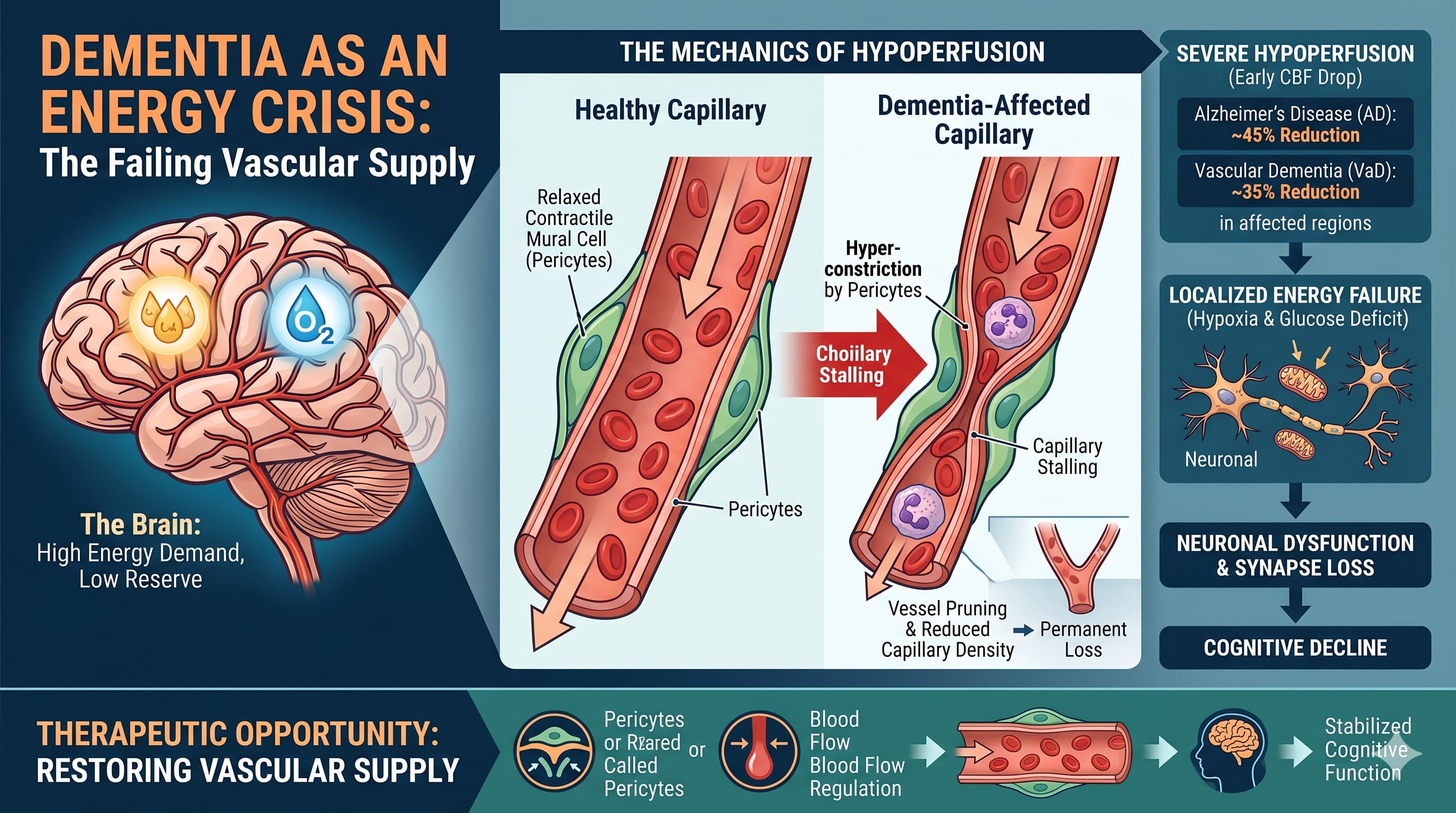
The most critical finding is the ISMN + any PDE5 inhibitor = absolute contraindication. If sildenafil is in the longevity stack, the LACI-2 combination is off the table entirely. These two cGMP-raising strategies cannot coexist.
The NAC problem: The 62% CBF improvement cited in the paper was IV, acute, at 50 mg/kg in mice (HED ~284 mg IV). Standard oral supplement doses almost certainly do not replicate this at the pericyte level. Oral NAC is defensible as a low-cost antioxidant, not as a validated CBF intervention.
The rapamycin interaction: Nimodipine is a moderate CYP3A4 inhibitor. For anyone on rapamycin, adding nimodipine requires blood level monitoring and likely a rapamycin dose reduction — this is a clinically significant interaction that gets no attention in the longevity community.
The best ROI in the stack: Sildenafil (25–50 mg daily). Observational human AD-prevention signal, RCT evidence in SVD, mechanistically coherent, 40% oral bioavailability, LD50 >7,500 mg/kg, and $15–60/month. The only non-negotiable: absolutely no nitrate drugs. Sourcing: Rx (or OTC in some markets). Generic tadalafil and sildenafil widely available. Tadalafil (longer half-life, once daily) may be preferable for a longevity protocol.
Sources consulted for this research include FDA prescribing information databases, PubMed pharmacokinetics literature, the LACI-2 and NILVAD trial protocols, and the Korte 2024 primary study.
Summary Comparison Table
| Drug |
Node Targeted |
Mouse Dose |
HED (mg/day, 70 kg) |
Clinical Dose |
Bioavailability |
Half-life |
Key CYP |
Key Safety Flag |
Monthly Cost |
Evidence Level |
| NAC |
ROS (upstream) |
50 mg/kg IV |
~284 mg IV |
600–1,800 mg oral |
4–10% oral |
5–6 h |
Minimal |
High-dose hepatotox |
$10–30 |
Mouse IV only |
| Nimodipine |
VGCC/Ca2+ (pericyte) |
~25 mg/kg/day p.o. (estimated) |
~1,580 mg/day (corrected for BA) |
90 mg/day clinical |
4–13% |
8–9 h |
CYP3A4 (sub+inhib) |
Rapamycin DDI; hypotension |
$50–100 |
Phase 2 human |
| Sildenafil |
PDE5/cGMP |
N/A (human data) |
N/A |
25–50 mg/day |
40% |
3–4 h |
CYP3A4/2C9 |
Nitrate contraindication |
$15–60 |
Observational + Phase 2 |
| Nilvadipine |
VGCC/Ca2+ (pericyte) |
N/A |
N/A |
8 mg/day SR |
14–19% |
11–20 h |
CYP3A4 sub |
Not FDA-approved in US |
$30–80 |
Phase 3 (failed primary) |
| ISMN |
NO/cGMP (eNOS) |
N/A |
N/A |
40–60 mg/day |
~100% |
5 h |
Minimal |
PDE5i contraindication; tolerance |
$10–20 |
Phase 2 (LACI-2) |
| Cilostazol |
PDE3/cAMP + antiplatelet |
N/A |
N/A |
200 mg/day |
Variable |
11–13 h |
CYP3A4/2C19 |
Heart failure CI; CYP DDIs |
$15–40 |
Phase 2 (LACI-2) |
Protocol-Breaking Interaction Alert
ISMN + Sildenafil = ABSOLUTE CONTRAINDICATION. These two drugs cannot be co-administered under any circumstances. A longevity protocol must choose one cGMP-raising strategy:
- Path A (SVD/NO-deficient): ISMN + Cilostazol (the LACI-2 combination)
- Path B (AD-predominant/pericyte): Nimodipine or Nilvadipine + Sildenafil
Combining nimodipine and sildenafil is pharmacodynamically permissible but carries additive hypotension risk. Blood pressure monitoring is mandatory.
THE STRATEGIC FAQ
Ten questions a longevity specialist would bring to the lead author, with answers drawn from the paper and external evidence.
Q1. The paper’s key pericyte mechanism is established almost entirely in transgenic mouse models overexpressing familial AD mutations. These represent less than 5% of all AD cases. How confident are you that sporadic late-onset AD, which is what 95% of patients actually have, operates through the same Abeta-pericyte-ET1 cascade?
A: This is the most significant translational gap in the paper. The AppNL-G-F model uses a humanized knock-in of familial APP mutations (Swedish, Beyreuther-Iberian, Arctic), which drive Abeta overproduction from endogenous promoters — still a better model than older transgenic overexpressors, but not sporadic AD. The pericyte constriction mechanism has partial human validation: postmortem brain tissue from sporadic AD patients shows reduced capillary diameter near pericyte somata (Nortley 2019), and applying Abeta oligomers at concentrations matching sporadic AD brain tissue to human cortical tissue constricts capillaries. The ET1/endothelin converting enzyme-2 elevation has also been confirmed in human sporadic AD postmortem tissue. So the mechanism has some human pathological support. However, whether a primary vascular trigger (rather than secondary Abeta elevation) initiates pericyte dysfunction in sporadic AD is entirely unresolved. In individuals without familial Abeta mutations, the upstream triggers may be hypertension, APOE4-driven BBB dysfunction, or aging-associated pericyte senescence rather than Abeta itself. The cascade may be convergent — different upstream inputs, same downstream pericyte contraction endpoint — but this has not been formally demonstrated. [Confidence: Medium that the downstream mechanism is shared; Low that the upstream trigger is identical in sporadic AD.]
Q2. The review states CBF falls 0.3–0.5% per year in healthy aging. At that rate, is there a quantitative CBF threshold below which cognitive impairment becomes irreversible, and does current evidence allow us to define a therapeutic window?
A: The paper identifies two functional thresholds from rodent studies: a 20% CBF reduction impairs attention, and a 30% reduction impairs spatial memory in rats. In humans, a 50% reduction causes loss of consciousness. For AD, the CBF deficit reaches 45–50% in affected areas — perilously close to the rodent threshold for severe impairment. Critically, pericyte-mediated capillary constriction becomes potentially irreversible once pericytes constrict and die, which the paper describes as a “semi-permanent” capillary diameter reduction. Post-stroke no-reflow is a clinical exemplar of this. The therapeutic window implied by the data is pre-clinical to mild cognitive impairment (MCI) — i.e., before significant pericyte death occurs. APOE4 genotype further tightens this window, as BBB and vascular changes begin in the 3rd–4th decade in APOE4 carriers. The paper explicitly argues for prophylactic treatment analogous to statin therapy for cardiovascular disease: start before symptoms appear. The biomarker trigger they propose is ASL MRI (CBF reduction) or DCE-MRI (BBB Ktrans elevation), but these are not yet in clinical routine. [Confidence: High that a window exists; Medium on exact timing; Low on validated clinical biomarker thresholds for treatment initiation.]
Q3. Capillary stalling by neutrophils is presented as a major mechanism. But the paper acknowledges that human neutrophils are 50–70% of circulating leukocytes versus 10–25% in mice. Does this mean all mouse stalling data systematically underestimates the human disease burden, and does this make anti-neutrophil strategies the most underrated therapeutic approach here?
A: Almost certainly yes to the first question, and possibly yes to the second. The quantitative disparity is stark: if 5% of capillary segments are blocked in AD mice (versus 0.4% in wildtype), and the human stalling driver — neutrophils — is 2–7x more prevalent and individually larger in human blood than in mouse blood, then extrapolating directly from mouse stalling data to human disease burden requires a scaling correction that has never been formally applied. Human capillaries are also larger than mouse capillaries, which partially offsets the neutrophil size problem, but net stalling burden in humans likely exceeds mouse models significantly. The therapeutic implication is that anti-adhesion strategies (targeting VCAM1/ICAM1 on endothelium, LFA-1 integrin on neutrophils, or P-selectin) may have a larger effect in humans than the ~26–32% CBF improvement seen in AD mice with neutrophil-binding antibodies. No anti-stalling agent is currently in clinical trials specifically for AD or VaD. This is a legitimate under-invested target. The paper notes that neutrophil depletion alone is sufficient to improve cognitive function and reduce Abeta1-40 in AD mice — a dramatic result that argues for therapeutic prioritization. [Confidence: High that stalling is underestimated; Medium on therapeutic impact magnitude in humans.]
Q4. Several of these drugs (nimodipine, ISMN, sildenafil) lower blood pressure as part of their mechanism. In an older population already at risk of orthostatic hypotension and falls, how do you balance the CBF-raising benefit against systemic hypotension risk — especially given the paper’s own note that excessive CBF reduction from impaired autoregulation during postural change is a problem?
A: This is an underappreciated hazard. The paper acknowledges that aging impairs pressure autoregulation, increasing the risk that any postural change (sit-to-stand) produces a transient cerebral hypoperfusion event. Adding vasodilating drugs to this physiological background increases that risk. The key clinical distinctions are:
First, nimodipine at 90 mg/day has a relatively modest systemic hypotensive effect in normotensive individuals due to its partial cerebrovascular selectivity. Its safety at this dose in 500+ patients over 18 months in the NILVAD trial was acceptable. Second, ISMN’s hypotensive effect is more pronounced and requires the nitrate-free interval strategy to manage tolerance and limit continuous blood pressure depression. Third, the combination of ISMN + cilostazol (two vasodilating agents) amplifies hypotension risk, which is why LACI-2 titrated doses slowly and monitored standing blood pressure. The practical protocol answer: titrate slowly, use the lowest effective dose, measure supine and standing blood pressure routinely, and avoid these drugs in patients with pre-existing orthostatic hypotension, baseline low blood pressure (systolic below 110 mmHg), or concurrent alpha-blockers. [Confidence: High that the risk is real; Medium on magnitude in carefully monitored longevity patients.]
Q5. The paper proposes arterial spin labeling (ASL) MRI as a biomarker to trigger treatment initiation. Is this ready for clinical use, and what is the minimum CBF reduction detectable and clinically meaningful on current ASL platforms?
A: ASL MRI is clinically available on 3T and 7T scanners and produces reproducible quantitative CBF maps without contrast agent. It is used in specialist centres and increasingly in clinical research protocols. However, it is not yet routine in primary care or general neurology practice. The practical barriers are scanner availability, scan time (10–20 minutes), and standardization across sites and field strengths. The sensitivity question is important: the age-related CBF decline of 0.3–0.5% per year is subtle enough that cross-sectional ASL comparisons to age-matched norms require careful correction for gray matter atrophy, blood pressure, and hematocrit — all of which confound the CBF signal. Serial scanning in the same individual (tracking rate of decline rather than absolute CBF) is more informative and analytically feasible but requires a baseline and follow-up design. The paper cites evidence that CBF reduction is detectable and correlates with cognitive impairment across 95% of brain regions in healthy older adults. The minimum clinically actionable threshold has not been formally defined — this is an active research gap. In practice, a reasonable clinical algorithm would be: APOE4 screening → baseline ASL MRI in 5th decade → repeat every 2–3 years → treatment consideration if decline exceeds 15–20% of regional baseline. This algorithm does not yet exist in any guideline. [Confidence: High that ASL is technically capable; Low that clinical implementation thresholds are established.]
Q6. Is there any human evidence that oral NAC at supplement doses (600–1,800 mg/day) actually reduces ROS in the brain microvasculature specifically — not just systemic oxidative stress? Without this, recommending NAC for pericyte-directed CBF improvement seems mechanistically unjustified.
A: This is the correct challenge. The published 62% CBF improvement in AD mice was produced by an acute IV injection of 50 mg/kg — a high-exposure, BBB-crossing route delivering a bolus systemic ROS scavenge. There is no published data showing that oral NAC at 600–1,800 mg/day achieves sufficient CNS penetration and local ROS reduction at the microvasculature to meaningfully suppress ET1 production from perivascular macrophages. One ASL MRI study in multiple sclerosis patients showed that IV NAC (150 mg/kg over 24h) increased cortical CBF by approximately 6–7%, suggesting some CNS vascular effect at high IV doses, but this is far from oral supplement doses. For oral chronic use, the primary measurable effects are plasma glutathione replenishment and systemic oxidative stress reduction (F2-isoprostanes, 8-OHdG). Whether this translates to reduced microglial NOX2 activity at the blood-brain interface is speculative. NAC-amide (NACA), a more BBB-penetrant acetylated form, may be a pharmacologically superior option for CNS-directed antioxidant effects, but it is not widely studied or commercially available. Bottom line: recommending oral NAC as a pericyte/CBF intervention based on the Korte 2024 data is premature. It is defensible as a low-cost antioxidant adjunct with a benign safety profile, not as a validated pericyte-directed CBF intervention. [Confidence: High that the mechanistic gap is real; Low that standard oral NAC doses replicate the IV mouse effect.]
Q7. Oligodendrocytes producing Abeta is mentioned as a new finding. If white matter oligodendrocytes are a local source of Abeta that drives capillary constriction specifically in white matter, does this mean that white matter and grey matter require different therapeutic strategies — and could VGCC blockers, which work in cortical grey matter models, miss the white matter pathology that characterizes VaD?
A: This is the most intellectually important open question raised in the paper, and the authors themselves flag it explicitly. The finding that oligodendrocytes contribute meaningfully to Abeta production in the white matter (Rajani 2024, Sasmita 2024) raises the question of whether there is a second, oligodendrocyte-driven Abeta-pericyte constriction cascade operating in white matter independently of the grey matter neuronal/microglial cascade. If so, the white matter vascular pathology in VaD and AD may have a cell-autonomous oligodendrocyte component that VGCC blockers, tested almost exclusively in cortical grey matter models, might only partially address. The white matter further has: lower baseline capillary density, downstream blood flow position, impaired CO2 vasoreactivity, and oligodendrocytes that are intrinsically more vulnerable to ischemia. A white matter-specific therapeutic approach — potentially targeting oligodendrocyte Abeta production directly, or using vasodilators with preferential white matter penetration — has not been developed. Acetazolamide increased white matter CBF in CADASIL patients 20 years ago (the Bousser group result cited in the paper), and adrenomedullin is now in CADASIL trials, suggesting white matter vascular targets are being explored. But the oligodendrocyte-Abeta-capillary link is too new (2024) to have informed any current therapeutic program. [Confidence: High that grey and white matter pathophysiology differ; Medium on the clinical implication for drug selection.]
Q8. The review emphasizes the APOE4 genotype as a high-risk profile for early vascular dysfunction. Should APOE4 homozygotes (2–3% of the population, ~12x higher AD risk) be placed on CBF-protective therapies prophylactically in their 40s? What does the evidence actually support?
A: The evidence supports early vascular intervention in APOE4 carriers — but “in their 40s” remains speculative without RCT data. The mechanistic case is strong: APOE4 carriers show early reductions in cerebral blood volume and capillary dysfunction (Aamand 2016), reduced cerebrovascular reactivity in young adults (Suri 2015), BBB breakdown in the hippocampus correlating with cognitive decline (Montagne 2020), and CSF PDGFRbeta elevation (pericyte injury marker) even before Abeta deposition. Taken together, these data strongly suggest that vascular dysfunction begins 1–2 decades before clinical AD in APOE4 carriers. What the evidence does NOT support yet: (1) a randomized trial showing that early CBF-raising therapy in presymptomatic APOE4 carriers prevents dementia; (2) identification of the safest agent for a 20–30 year prophylactic protocol; (3) the optimal initiation age. The closest real-world parallel is statin therapy in high-risk cardiovascular genotypes: we accept prophylactic treatment before clinical disease because the pathophysiology is understood and the drugs are safe. The same reasoning applies here — but the drugs are less well-characterized for multi-decade use than statins. For an APOE4 homozygote aged 45 with normal cognition and a family history of early AD: the mechanistic argument for low-dose nimodipine or sildenafil (given their safety profiles) combined with aggressive blood pressure optimization is defensible. No guidelines endorse this yet. [Confidence: High that early intervention is biologically rational; Low that any specific protocol is proven.]
Q9. How does restoring CBF interact with current immunotherapy approaches targeting Abeta (lecanemab, donanemab)? Could CBF restoration actually increase Abeta clearance sufficiently to compete with immunotherapy, or are these complementary?
A: The mechanisms are complementary, not competitive. Immunotherapy (lecanemab, donanemab) works by promoting microglial-mediated clearance of existing Abeta aggregates and plaques. CBF restoration works upstream by: (1) reducing hypoxia-driven BACE1 upregulation (less new Abeta produced); (2) improving vascular clearance of soluble Abeta via LRP1 transport across endothelium and via spontaneous vasomotion driving perivascular drainage; (3) reducing the endothelial inflammation that promotes neutrophil stalling and further CBF reduction. Interestingly, a 2025 study cited in the paper (Rajani 2024) showed that ASL MRI CBF increased after lecanemab infusions in AD patients — direct evidence that Abeta clearance by immunotherapy restores some CBF. This supports the bidirectionality of the Abeta-CBF relationship: Abeta worsens CBF and CBF reduction worsens Abeta. Combining a CBF-restoring agent with immunotherapy could theoretically enhance the therapeutic effect by: reducing the rate of new Abeta generation while immunotherapy clears existing aggregates; and improving cerebrovascular health to sustain long-term benefit. No combination trial has been done. The key safety consideration for combination: amyloid-related imaging abnormalities (ARIA) — microbleeds and edema that occur with immunotherapy — might be exacerbated by vasodilating agents that increase perfusion pressure in already-compromised vessels. This combination requires careful monitoring. [Confidence: Medium-High on complementarity; Low on safety of combination.]