A Gladyshev-lab computational study has built the first aging clock derived from RNA-level damage signatures — not gene expression — showing that transcriptomic deterioration accumulates predictably with age, is partially reversible by rapamycin and caloric restriction, and is measurably accelerated in Alzheimer’s disease blood at a signal strength that conventional expression-based clocks completely miss.
Aging researchers have argued for decades that molecular damage — not just regulatory drift — is a root cause of biological decline. The evidence base for DNA mutations and protein oxidation is well established. But one entire layer of molecular deterioration has escaped systematic quantification: the damage accumulating in RNA itself.
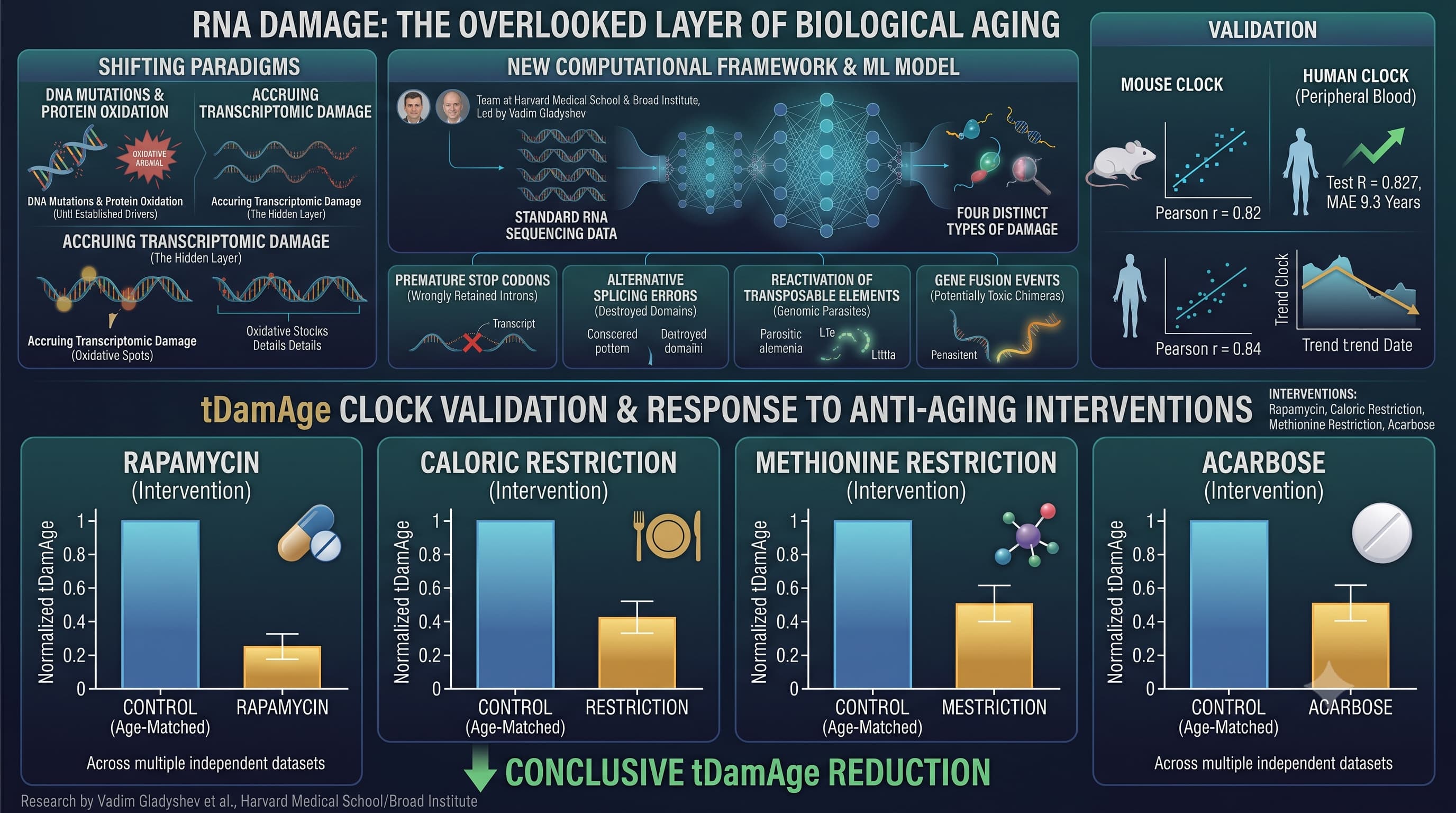
A team at Harvard Medical School and the Broad Institute, led by Vadim Gladyshev, has now built a computational framework that extracts four distinct types of transcriptomic damage from standard RNA sequencing data. These are not soft correlations. The damage types are structurally concrete: premature stop codons generated when introns are wrongly retained in transcripts (producing truncated, non-functional proteins or triggering degradation); alternative splicing errors that physically destroy conserved protein domains; the reactivation of transposable elements — genomic parasites that are normally suppressed — across multiple repeat classes; and gene fusion events, where unrelated RNA strands are incorrectly joined into potentially toxic chimeric molecules. Each of these increases with age. All four damage types positively correlate with chronological age across the majority of human and mouse tissues tested.
Using the GTEx dataset — over 16,000 human tissue samples from 26 tissue types — the team confirmed consistent age-related accumulation across nearly every tissue. The cerebellum carries the highest damage burden of any brain region, consistent with its documented vulnerability to aging-related neurodegeneration. Pancreas, muscle, and blood show the least damage, potentially reflecting higher cell turnover that clears damaged transcriptome-burdened cells before accumulation peaks.
From these signals, the team trained a machine learning aging clock called tDamAge — first in mice (Pearson r = 0.82) and then in human peripheral blood (test R = 0.827, mean absolute error 9.3 years). The clock responds to interventions in the expected direction across multiple independent datasets: rapamycin, caloric restriction, methionine restriction, and acarbose each reduce tDamAge relative to age-matched controls; SARS-CoV-2 lung infection, BubR1 progeroid mutation, and Klotho heterozygous knockout elevate it.
The most clinically striking finding is in Alzheimer’s disease. When the human blood tDamAge clock was applied to AD patient samples, it detected significantly elevated biological age acceleration compared to cognitively normal controls (p = 0.0099). A standard gene expression clock — built from the same datasets using the same pipeline — failed to detect any significant difference. Transcriptomic damage picks up a signal that expression levels obscure.
The paper also captures a transient rejuvenation window during mouse embryogenesis — a pronounced drop in tDamAge from approximately embryonic day E10/11 through E16 — with the damage clock’s nadir arriving slightly later than expression-based clocks, suggesting damage clearance lags transcriptional reset during development.
The authors frame this within Gladyshev’s deleteriome theory: that aging is defined by the totality of accumulated molecular damage across biological layers. tDamAge provides the first scalable, RNA-layer readout of that entropy. That framing is theoretically sound, though the paper stops short of formal causal proof — a distinction worth keeping in mind.
Actionable Insights
For the clinician and advanced biohacker, the intervention-response data is the most practically relevant output. Anti-aging interventions already in widespread use — rapamycin, caloric restriction, acarbose, and methionine restriction — each produced measurable reductions in transcriptomic damage age in mice, with fold-change reductions estimated at approximately 5 to 30 percent relative to age-matched controls depending on tissue and age stratum (from Figure 4H). Methionine restriction and dietary restriction showed the most consistent reductions across tissue types; rapamycin’s effects were meaningful but more tissue-specific.
Convergence analysis across 22 longevity intervention datasets identified RNA splicing fidelity, chromatin organization, and RNA catabolism as the central shared targets — suggesting these aren’t incidental effects but core mechanistic pathways through which diverse anti-aging strategies operate.
The AD blood signal (approximately 2 to 4 years of tDamAge acceleration relative to controls, estimated from Figure 6F) positions peripheral blood tDamAge as a potential early neurodegeneration marker. At current accuracy (MAE approximately 9.3 years), this clock is unsuitable for individual-level biological age tracking but may have utility as a population-level stratification tool.
Source:
- Open Access Paper: Causally measuring aging and rejuvenation through transcriptomic damage
- Institution: Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Broad Institute of MIT and Harvard, Cambridge, MA, USA
- Country: USA
- Journal: bioRxiv preprint; posted June 29, 2026. Not peer-reviewed at time of analysis.
- Impact Evaluation: This work is a preprint and carries no journal impact factor.
Related Reading:
- Lost in Translation: Why Your Cells Mis-Read Their Own Genes As You Age — And How Rapamycin May Fix It
- Rapamycin exerts geroprotective effects in the ageing human immune system by enhancing resilience against DNA damage
- Rapamycin Clears Alzheimer's Plaques by Rebooting Microglial Fat Metabolism—But Females Benefit Most
- A Tale of Two Sexes: Rapamycin and Friends Protect Bone in Females, Lifespan in Males
- Short-term rapamycin treatment improves embryo quality, pregnancy, and live births