Very interesting. Great presenter. Will definitely have to check out some of his other things.
This is certainly interesting.
So the baseline CAC score matters and progression > 15% per year matters a great deal. This seems to be the case even in the face of low LDL levels.
Maybe LDL is a poor marker compared to some other measures. Maybe some people are so genetically prone to CVD that nothing much stops the progression.
The score certainly has value and is underutilized.
2 Likes
ABSOLUTELY, something found in repeated future studies (this study was in 2004)
And yes, that’s why a first MI is found in high and low LDL persons.
That’s a more interesting question…but anyone interested in living longer needs to be checking this marker, and intervene with whatever is in the toolbox (lifestyle, meds).
And there must surely be a genetic component, beyond just classic FH or LPA genes.
The long lived, they have some special CVD deferral genes, this is widely shown in GWAS studies.
1 Like
Yes, the long lived have a deferral mechanism for basically everything. It’s like they’re on an intrinsic mTOR inhibitor.
They’re healthy and avoid major illnesses for most of their life and then suddenly get something and fall off the cliff.
That’s why I don’t separate healthspan and lifespan the way others seem to do. They’re very related.
3 Likes
I was aware of this paradox but not that statins do the same.
Human mole rat equivalents, who age epigenetically but not by classic markers. Then just keel over at end of life. Lost a couple of relatives near 95 yr old. Got sick and quickly passed away.
I totally agree. Longevity, medically extended, with major comorbidities, reduced independent living, partial cognitive, is not a “lifespan” target. Just a longevity statistic.
Re statins increasing CAC score:
1 Like
So several studies have shown the benefits of pycnogenol with Centella A. in reducing atherosclerosis and CAD. It would appear that plaque stabilization is also a feature of Centella.
Totally agree.
That’s what I want to be , a human naked mole rat where my probability of death never increases.
It’s a point that I’ve heard several times that in the study population of say 12 million, maybe 4 million of them have an underlying silent cancer driving down their LDL. Many of these cancers that are silent wouldn’t kill them, or at least not for many, many years, like a Gleason 6 prostate, very well differentiated breast, non melanoma skin, etc.
The most common bowel disease is irritable bowel syndrome and that’s not increasing mortality.
Also, let’s say a drug like a statin works through several mechanisms, so maybe its anti inflammatory and anti cancer effects negate the increase in mortality from lowering the LDL.
1 Like
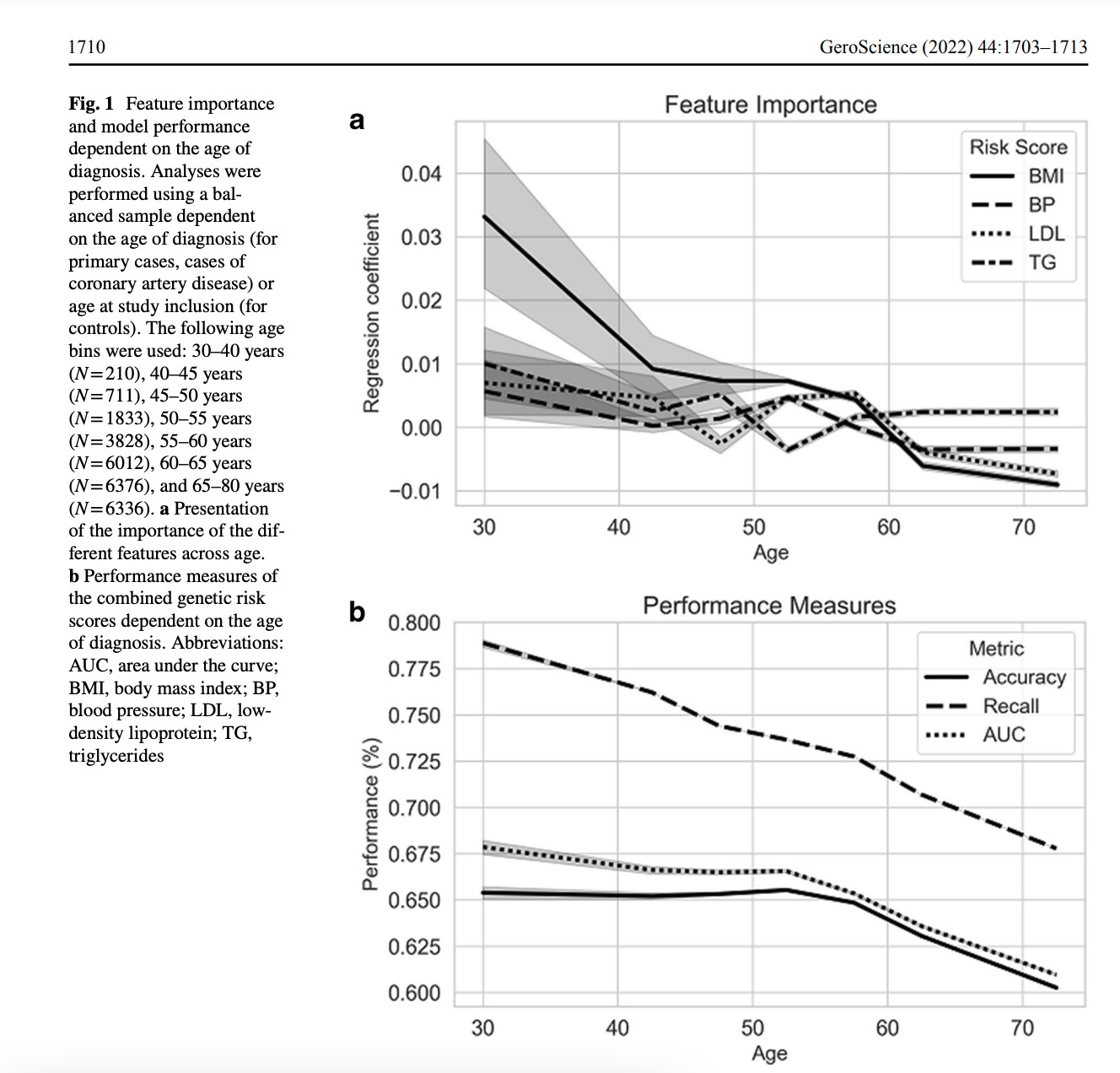
Just in. A Mendelian randomization study on coronary risk factors as they relate to age. None of the classical risks are significant after the age of 70.
I thought I had read that a few times in passing, so had a look:
Conclusion: Low plasma LDL cholesterol levels were robustly associated with an increased risk of cancer, but genetically decreased LDL cholesterol was not. This finding suggests that low LDL cholesterol levels per se do not cause cancer.
“Low LDL cholesterol in patients with no history of taking cholesterol-lowering drugs predates cancer risk by decades, suggesting there may be some underlying mechanism affecting both cancer and low LDL cholesterol”
Classic association does not mean causation confounding bias.
1 Like
It’s fascinating to say the least. So low LDL predates cancer by Decades but there appears to be a common denominator here.
What if the same genetic locus is predisposing to Both cancer and low LDL. The LDL factor is showing up first. Cancer is taking longer to develop. Once it does develop it consumes LDL , lowering it even further.
Now enter rapamycin. Rapamycin slows cancer cell proliferation resulting in less LDL consumption and the levels start to increase ,even in people with an underlying malignancy.
Interesting theory.
From the paper:
“How preclinical cancers might cause low plasma LDL cholesterol levels is not known. Potential mechanisms include effects on cholesterol absorption, transport, metabolism, or utilization. In support of the notion that preclinical cancers increase LDL cholesterol metabolism or utilization, previous studies have shown that plasma cholesterol levels are inversely associated with tumor mass of hematological cancers and that plasma cholesterol levels revert to normal after cancer remission (40,41). This idea is also supported by the specific cancer subtypes that have been observed to be associated with low LDL cholesterol levels in this and previous studies, that is, gastrointestinal cancer, hematological cancer, female-specific cancers, urological cancer (1,2,4), and lung cancer (2,4). All of these cancers have the potential to produce large tumors and metastases and can remain preclinical for a considerable time before diagnosis”
Some confounders that cause both lower lipids and cancer…smoking, alcoholism, COPD also reduce lipids, associated with cancer.
In rapamycin/mice studies, rapamycin administration is not associated with increasing lipids, even though it defers cancer?
Is it possible the cancer and lipids are distinct but yet dually mediated mechanisms re rapamycin? Rapamycin reduces mTOR expression in cancer cells, and at the same time, is causing a rise in lipids via liver expression, as we see very commonly see in humans, even the cancer cohorts? Most certainly in the human cancer cohorts, lipids do NOT go down (only up) with rapamycin, even with though supposed reduced cancer proliferation (the cancer/GFJ study only reported sirolimus pharmacokinetics, mTOR, and “stable” disease/progression, not any cancer specific additional markers).
Perhaps rapamycin is a “net” effect…reduced LDL by the reducing cancer “signal”, but net higher via liver mediated LDL?
The “escapers”.
“Our analyses suggest that the classical primary CAD risk factors predominantly affect those at young age. Subsequent stratification based on cardiovascular disease history revealed that the pravastatin-induced decrease in vascular event rate was only observed in those with a vascular event in their history [8]. This suggests treatment with cholesterol-lowering therapy does not yield clinical benefit on primary CAD prevention in older people.”
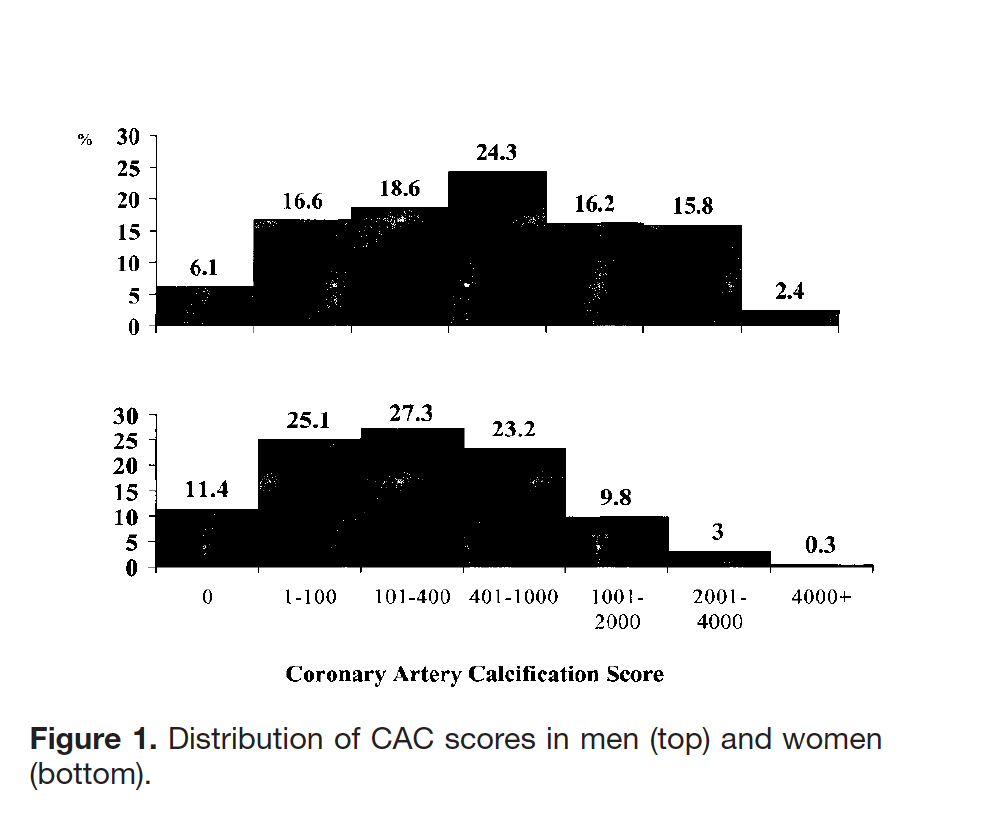
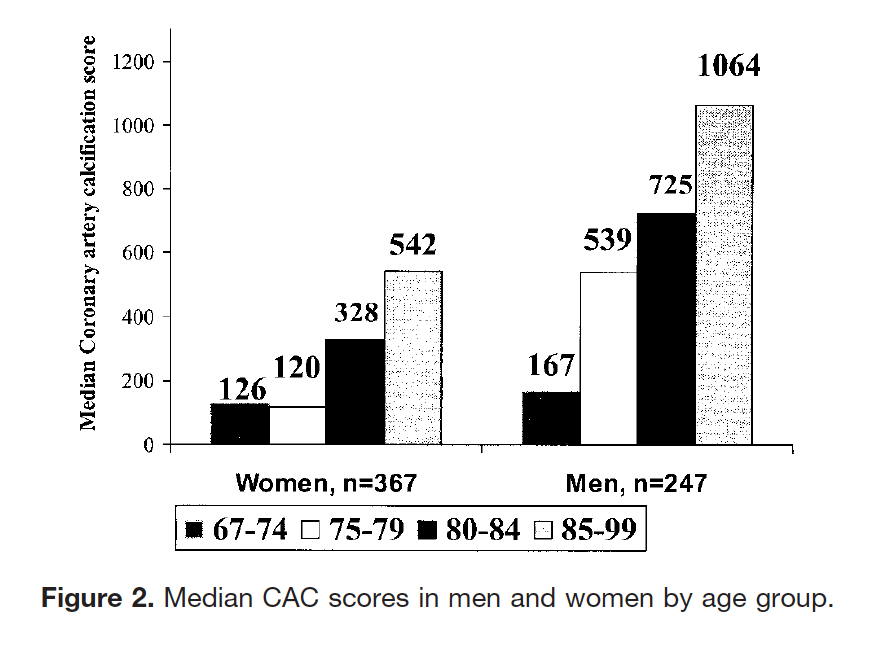
And it’s NOT because they have LOW CAC scores.
Coronary Artery Calcification in Older Adults to Age 99 Prevalence and Risk Factors
https://www.ahajournals.org/doi/epub/10.1161/hc4601.099464
“The key findings of this study were that the extent of coronary artery calcification was strongly associated with age through the ninth decade in men and women and was associated with CVD. Second, risk factor levels were only weakly associated with the extent of CAC in these older adults.”
Conclusions—A wide range of CAC scores was observed, suggesting adaptation with aging. CAC may have potential to predict CVD in older adults, but this remains to be determined.
1 Like
I don’t know about you, but I now have some comfort knowing that my lipids are a little on the high side. Haha
It would appear that the CAD risk factors are mostly prognostic in a person’s middle age, basically under 50.
So let’s get back to the original topic. You’ve been good with your lipids and then you started rapamycin at say 65 or older, which probably represents the majority of users, and your lipids take a jump.
Do you intervene? Is it a risk?
Doesn’t look like it.
3 Likes
Well, you just put in a big caveat…you hadn’t before I don’t think.
What IF you have high lipids, have other CVD risk factors, especially a higher % CAC score, and your lipids rise taking rapamycin in an attempt to reduce your CVD risk? And “lipids take a jump”, that’s yet another covariate, rise by how much, and which lipid(s)?
For good lipids, low CAC, no other CVD risk factors…me personally (age, health, risk factors) with what I know at time of writing, I probably would not. But…I’d probably get a more advanced atherogenic risk panel done to look deeper in the meantime as hedge. And probably check CAC again in a year (setting aside the radiation exposure risk!)
So I’ll rephrase.
My main point all along has been whether we should react to an elevation in lipids From rapamycin and intervene. The assumption here has been that lipids haven’t been a concern as a CAD risk factor until rapamycin was started, at which point the levels went up.
I still don’t think it’s a concern.
2 Likes
I haven’t accepted benevolence per Dr B. I would not treat, but increase vigilance/monitoring.
In fact, I haven’t accepted anything re Rapamycin in humans with our current dosing regimes and inability to monitor at deeper tissue levels. But I’m a rowing team member!