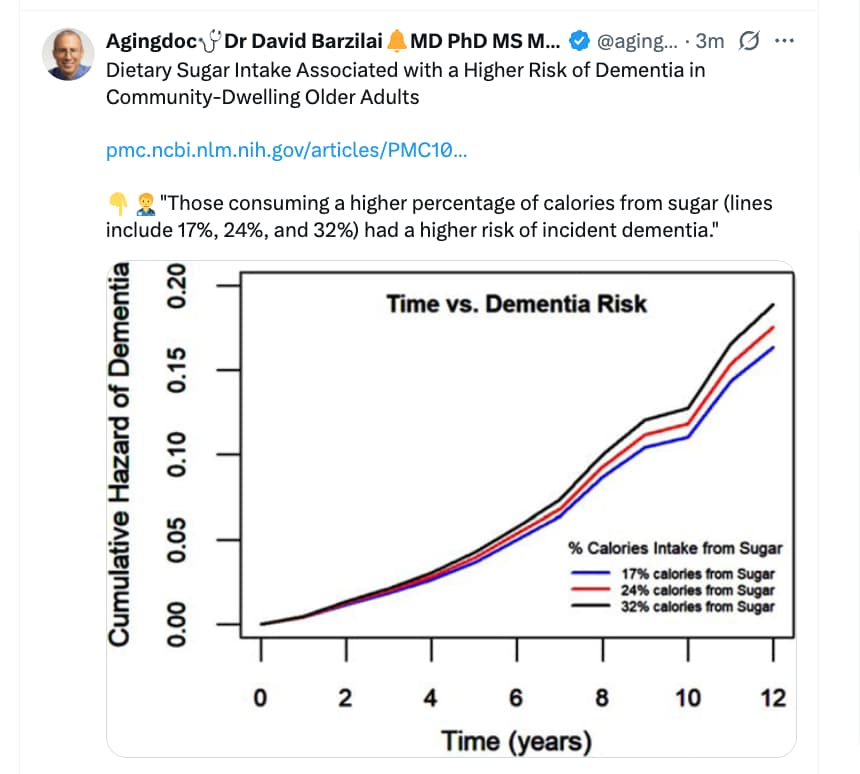
Okay Attia is right, diet studies are absolute garbage, and can tell you whatever you want to hear.
Not that some studies aren’t that bad and some ideas. But believing dietary sugar is causing twice the rate of dementia on its own with no residual confounding or mediating factor just feels ridiculous to me.
I think you have to approach nutrition studies with a skeptical eye but it doesn’t make them garbage.
Does sugar intake track with dementia risk - yes. Is it causal? - I think so.
All we can really do is make observational studies but that doesn’t make nutrition less important. I can see the knee jerk reaction is to say we can’t study it well so we don’t know anything - so just forget it. But the relative importance of something doesn’t track with the ease of studying it.
Like how the general population doesn’t know whether they should eat eggs or not. Actually, no one knows - but we do know somethings even if we can’t know things with 100% certainty.
I personally think sugar intake causes dementia and the person with a failing brain eats more sugar. So the study will exaggerate the effect. We do know with near 100% certainty that diabetes and level of control of DM correlates very well with dementia.
At least this topic isn’t particularly controversial - eating excess sugar is bad.
But that’s not because of dietary sugar. That’s excess calories fattening up the pancreas above a specific fat threshold leading to T2D, as far as I’ve heard. Whether it’s sugar, or fatty meat it doesn’t matter. The problem is suggesting something is causal when it’s mediated by something else that has multiple salient factors. One might be hyperpalatable food.
To be fair the study claimed association, but we should maybe only link these studies when we think it’s causal, and specifically what, IMO.
@DrFraser I recently read that your post about LYSOVETA (the LPC form of omegas in the Accentrate product), so I thought I’d post what I found incase this is helpful to you or anyone else.
Dietary phospholipid carriers of DHA do not increase brain DHA levels: a replication study
“However, a subsequent study using a higher dose of sn -1 LPC-DHA did not confirm these findings and reported no significant increase in brain DHA”
https://www.jlr.org/article/S0022-2275(25)00175-0/fulltext
Because I learned about the importance of an APOE4 carrier to get DHA into the brain, and how challenging or impossible it might be, I have been in incessantly googling about how LPC omegas in Accentrate can do just that. (My goal was to make sure there was no other viable option because taking a fish product would be a big step for the vegan… I’m morally ok with doing it for health if there is no other option, even if mentally difficult)
In that effort, I found this replication study that questions if LPC does in fact do anything special at all… having said that, I’m not well versed at reading studies, so there could be nuance here that I’m not getting.
And if this study does in fact show that the LPC form doesn’t get into the brain any better, do you think this means that polar lipid forms of Algae Omega are not any better than non polar lipid forms?
I ask that becasue I’ve found that if polar lipids do help, ORLO might be one of the best brands for algae oil. If polar lipids don’t actually improve the bioavailability, then it seems the new Blueprint algae omega is most likely the best option with their much higher dosing.
(If Accentrate is still superior, as far as my personal problem goes, I realized I can most likely put their soft gels into an empty gel cap to hide the taste, but then I just have to worry about a potential fish burps… tba)
EDIT: OOOOHHHH… I keep looking to see what I’m missing here. Is it that the LPC form makes no difference if you don’t have apoe4, but perhaps this study is not relevant for someone with APOE4 and LPC is still the only way … and then maybe algae oil, even in polar lipid form is a waste of time??
On the useless of DHA supplementation see also:
Sure, but glucose eaten raises glucose in the blood which is the opposite of glucose control. So while your point could fully explain why DM (caused by meat consumption or excess calories etc), it can’t (IMO) explain how control of sugars reduces progression.
And since the meds reduce the progression to AD, it stands to reason that lower blood sugar is the reason. It would seem unlikely that several classes of drugs have effects independent of blood sugar control when it comes to AD prevention. I mean possible for 1 or 2 classes but not likely for all.
But, nothing 100%.
That’s temporary and the area under the curve is lower than for T2D. Of course suboptimal blood glucose destroys organs, arteries and veins. But I don’t see the point in arguing about this if it was not the study hypothesis. And I was aware that there was either confounding or mediating factor, that’s my point. It can be unhealthy user bias, excess calories leading to T2D (i.e T2D), blood glucose levels, etc, a more important factor than dietary sugar for us. If you believe dietary sugar is causing lots of dementia when most is actually some other factors, T2D, or excess calories leading to T2D, that’s bad.
Ok, I came at this with what I thought was well established science - sugar intake increasing risk of dementia - but just because I think it is true does not mean everyone agrees.
Certainly, DM is a risk factor. And my way of thinking is that we are all diabetics, just not yet crossing threshold. Now that is overly simplistic but is my way of thinking.
Maybe it’s because I bumped my A1C despite lowish BMI, heavy exercise and a low carb diet with lots of fiber and rare processed carbs. Now I am normal on no meds but I figure that I have one toe in DM.
Since medical school, I have always looked at sugar as toxic to all cells at sufficient levels. Necessary but toxic at a certain - easily crossed - point.
I think there is all sorts of evidence to back this up. Doesn’t make it true of course. But all studies are going to be confounded.
I believe you are not in the US and dietary studies in various countries are another huge source of confounding. In the US, the is a lot of HFCS which is felt to be significantly worse than sugar. So I would expect that US “sugar” is worse than EU or China sugar.
References 7-14 in this article link sugar intake to cognitive function or dementia in multiple populations.
Of course, we will never separate sugar intake from other unhealthy dietary choices in a population. Similar to saturated fat so we go based on what we have.
From 2 months ago .,.
https://www.sciencedirect.com/science/article/pii/S2274580725002547
The problem isn’t the sugar intake, it’s the serum glucose levels, and I think specifically insulin resistance, but I don’t know so much about this. Yes, serum glucose causes dementia. This is validated in mendelian randomization studies as well.
If an endurance athlete pours down sugar down his throat and goes on the bike, his serum glucose will barely move. He’s not increasing his risk of dementia, because he is insulin sensitive and using it.
I’ve seen anecdotes of people eating keto diet having prediabetes still. The cure is probably losing fat in the pancreas. People have differing levels of fat thresholds, people with a low BMI can still have a low threshold that causes T2D diabetes for them that’s not a problem for someone else. https://www.youtube.com/watch?v=epgRw-vIRSA
Saturated fats is not the same thing because on average that causes the diseased biomarkers, while sugar doesn’t have to but can. It’s hyperpalatable food causing weight gain that’s the problem, which includes sugar mixed with other foods.
in practice ectopic fat is deposited in all 3 major organ compartments (muscle, liver and pancreas). muscle insulin resistance (IR) generally sets in long before full blown diabetes. e.g. most obese people are insulin resistant but most aren’t diabetic. liver IR also generally precedes diabetes. initially, insulin resistant individuals are normoglycemic, as the pancreas is able to compensate by overproducing insulin. eventually, both pancreatic functional decline and liver insulin resistance (which causes uninhibited glucose production) result in hyperglycemia. there are many details but the gist of it is that ectopic fat accumulation causes organ dysfunction with accumulating metabolic impairment (IR, then prediabetes, then diabetes) – Gil Carvalho
The discussion on Omega 3’s continues to evolve; and each time I come up with a plan, I have to keep modifying it.
Dr. Kevin Tran does a good job discussing this and has pretty much an identical view to mine. We might both be wrong. Here is the link to his video.
Here is my current protocol … and I suspect it’ll change. I also think that using Doxycycline low dose with its activity on MMP-9 may be a big help.
ApoE4 carriers have a less efficient DHA transport system into the brain. This means that simply taking standard fish oil or DHA supplements may not provide the brain with sufficient levels.
Phospholipid-bound DHA (e.g., from krill oil) can help bypass the traditional DHA transporter.
However, much of the phospholipid structure may be cleaved during digestion, limiting how much actually reaches the brain.
ApoE4 heterozygotes (one copy of ApoE4) have a lower risk of deficiency compared to homozygotes (two copies), who may need higher intake and more targeted supplementation strategies.
Key Principles
Diversify your sources – Don’t rely on just one form of omega-3.
Small amounts go a long way – Only modest DHA levels in the right form are needed for the brain.
Aim for an Omega-3 Index of 8–10% – This supports heart and vascular health, while ensuring at least some DHA crosses into the brain.
Current Best Approach (subject to future updates)
Wild-caught fish as the foundation
Eat 4–6 oz of wild salmon a few times per week. Other excellent options: anchovies, sardines, mackerel, or sea bass.
Avoid large predator fish (e.g., tuna, swordfish) and farmed fish due to higher contaminant risk.
Source recommendation: NWWildFoods.com (use code wellness25 for a discount). Their wild berries and mushrooms are also excellent additions.
Krill oil for phospholipids and astaxanthin
Naturebell Antarctic Krill Oil – 2 capsules daily
240 mg EPA
160 mg DHA
400 mg Phospholipids
800 mcg Astaxanthin
Consider lysophosphatidylcholine (LPC) DHA for enhanced brain delivery
Accentrate Omega Max – 3 capsules daily
210 mg EPA
105 mg DHA
390 mg Lysophosphatidylcholines (LPCs – designed to cross the blood–brain barrier more effectively)
Accentrate link Omega-3 Supplement | Accentrate® Omega Max | Fenix Health – Fenix Health Science Discount code: GEF20 ($20 off)
Bottom Line
Start with fish first.
Use both krill oil and Accentrate to cover different delivery pathways.
Maintain a balanced Omega-3 Index (8–10%) to support cardiovascular and brain health.
Of note - on those codes … I make nothing … those are purely discount codes for my patients.
Based on what? Are there clinical trials on humans that are convincing? I feel like it’s just all mechanical assumptions based on longitudinal studies and the good old saying that “eating fish is good for your brain”.
I’m not diabetic, but my protocol with SGLT-2 inhibitors/ Empagliflozin is the following:
-
Supplement daily with D-mannose +Cranberries which can reduce the risk of UTIs: Combination of cranberry extract and D-mannose - possible enhancer of uropathogen sensitivity to antibiotics in acute therapy of urinary tract infections: Results of a pilot study - PMC
-
take the full 25mg dose about 1hr before a large meal which includes carbs / before consuming anything sugary. This tends towards daily when I’m traveling.
-
25mg/ 12.5mg / 0mg on other days based on how my CGM readings look. If my fasting / postpranadial glucose readings look satisfactory to me, then 0mg. If they’re a bit high, then 25mg/12.5mg depending on how unsatisfactory things look.
My hope is that this regimen minimizes my UTI risk while still providing the many benefits of SGLT-2 inhibitors.
I wouldn’t dream of stopping/pulsing SGLT2i based on abstract UTI fears, unless there’s a specific reason - I paused my empa because of surgery (recommended) and catheter abrasions. After a break I used urine test strips to establish that I likely am not harboring any bacteria in my track, and resumed empa.
Look at the literature. If you are a man, especially circumcised man, and not diabetic, you have essentially the same risk of UTI as someone who is not on an SGLTI.
Given this, I prefer to get continuous benefits of my empagliflozin. Of course I maintain good hygiene and also use urine test strips 1-2 times a month to spot any possible, however remote asymptomatic infections. YMMV.
I hadn’t thought of using urine test strips — gonna be using them!
Brain Stimulation Device Promising in Early Alzheimer’s Disease
A home-based brain stimulation device appeared to boost cognition in patients with early Alzheimer’s disease (AD), although the effect was small, a new study showed.
Patients who received active treatment with the device, which provides active gamma transcranial alternating current stimulation, showed improvements on several cognitive measures, including remembering faces and people’s names, said study author Barbara Borroni, MD, Molecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy.
“The effects we observed were modest but meaningful in everyday life,” Borroni told Medscape Medical News. “For example, patients showed improvements at being a bit quicker in recalling information or managing simple day-to-day tasks more independently.”
The study was reported on December 8 in JAMA Network Open.
This is additional evidence for gamma frequency stimulation with AD and there is also some evidence for PD. We personally use the Symbyx Neuro Helmet. It is relatively low cost as compared to other devices and seems to be a no-brainer to use - well a hope for avoiding no-brain.
I see this as a speculative therapy, where risk of harm is extremely low (apart from $1100 spent) and the benefit is likely. Hitting things from 10 different angles when dealing with neurocognitive decline risk seems sensible.
Multiple items will fail to be proven, but others will be (hopefully). We just can’t fully predict this, and the clock is ticking. I continue to see statins, ezetimibe, telmisartan, SGLT2-i, GLP-1, Omega 3 index, probably Vitamin D optimization, B12 optimization, low dose lithium, low dose doxycycline, optimizing insulin resistance, Blood pressure and probably a few other items as being important to longevity and to neurocognitive decline.
Yes a typo, thx for picking that up.
What is your view of PDE-5 inhibitors such as tadalafil to improve blood perfusion? And, what about intra-nasal insulin?