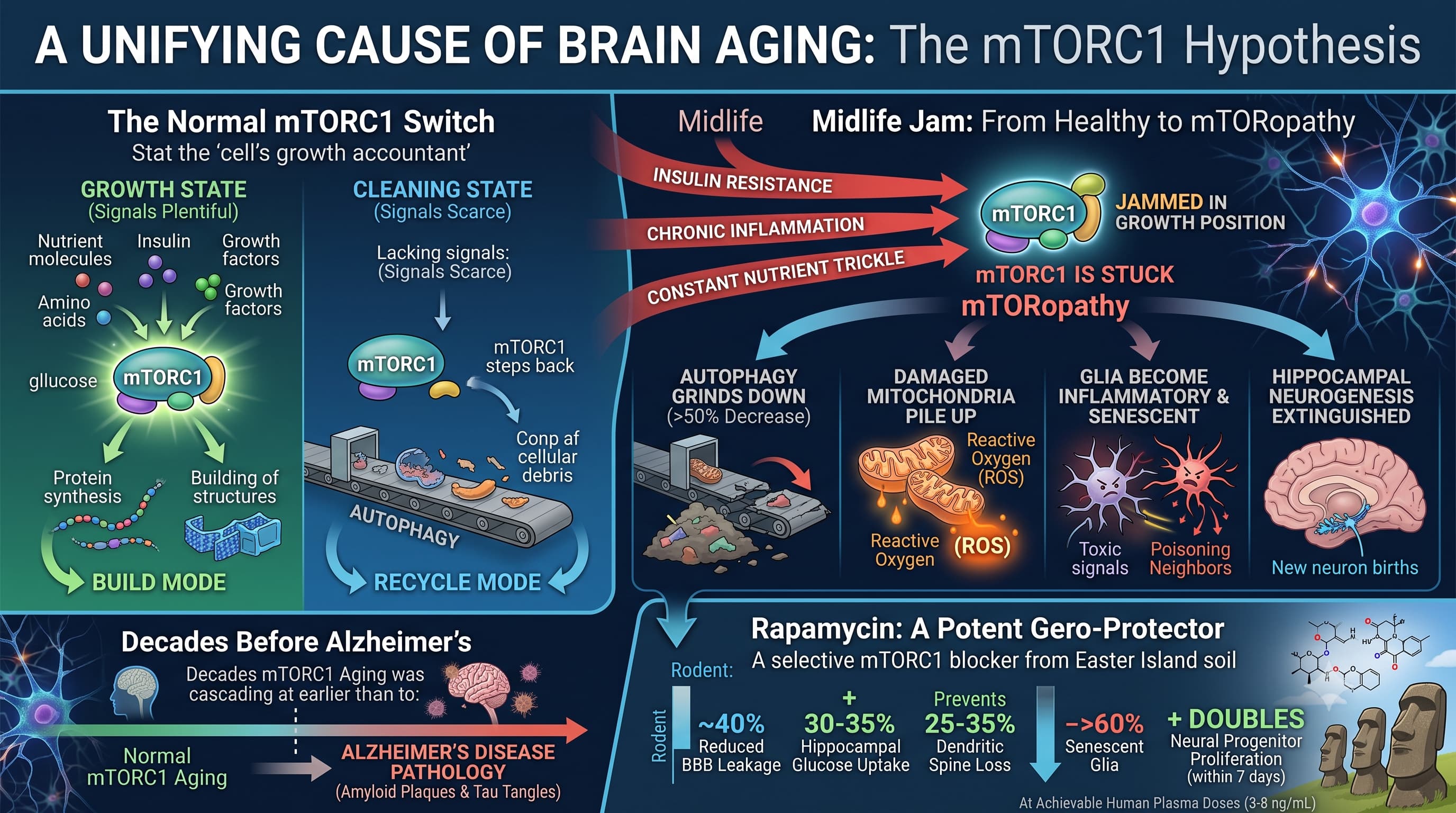
This review argues that most of the slow, universal decline of the aging brain traces back to one molecular culprit: chronic overactivity of mTORC1, a nutrient-sensing protein complex the authors nickname “mTORopathy.” When mTORC1 is stuck in the “on” position from midlife onward, it shuts down cellular housekeeping (autophagy), poisons mitochondria, inflames glial cells, and strangles the birth of new neurons. The authors marshal a large body of rodent evidence that intermittent, low-dose rapamycin — the drug that inhibits mTORC1 — can reverse, not merely slow, these changes even when started in old age.
For decades, neuroscientists have been unable to name a single unifying cause for the ordinary decline of the aging brain — the fading memory, the slower processing, the shrinking of connections that eventually touches nearly everyone who lives past 75. This review makes an ambitious claim: there is a common cause, and it is a protein complex called mTORC1.
mTORC1 is the cell’s growth accountant. When food, insulin, and growth signals are plentiful, it tells the cell to build; when they are scarce, it steps back and lets the cell clean house. The authors’ “big idea” is that from midlife onward this switch gets jammed in the growth position — by insulin resistance, chronic inflammation, and a constant trickle of nutrients — and never fully turns off. They call this stuck state mTORopathy.
The consequences, they argue, cascade through every compartment of the brain. Cellular recycling (autophagy) grinds down by more than half. Damaged mitochondria pile up and leak reactive oxygen. Support cells called glia slide into an inflammatory “senescent” state and poison their neighbors. The birth of new hippocampal neurons — a process now known to continue into old age in humans — is nearly extinguished. Crucially, the authors say all of this begins decades before the amyloid plaques and tau tangles of Alzheimer’s, meaning it is a feature of normal aging, not just disease.
The therapeutic hook is rapamycin, an immunosuppressant discovered in Easter Island soil that selectively blocks mTORC1. In aged mice, dogs, and marmosets, short intermittent courses reportedly restore blood flow, memory, synapse density, and neurogenesis to near-youthful levels — with benefits lasting months after the drug is stopped. The authors frame this as the single most mechanistically justified anti-brain-aging strategy currently available, and argue it outperforms every rival geroprotector tested (metformin, senolytics, NAD+ boosters).
The honest counterweight, which the authors do concede, is that this entire edifice rests on inbred lab rodents and monkeys living in sterile cages. No large human trial with cognitive endpoints has been done. There are no validated biomarkers to even measure brain mTORC1 in a living person.
Actionable Insights
The take-home messages are indirect, because the star intervention (rapamycin) is prescription-only and unproven for longevity use in humans. What the review supports, without needing a prescription:
The most robust, human-relevant lever is suppressing chronic mTORC1 activation through lifestyle modifications, because the review states that every well-validated geroprotector (caloric restriction, exercise, metformin, resveratrol, intermittent fasting) works largely by activating AMPK, which inhibits mTORC1. Magnitudes the authors cite for the drivers you can modify: midlife type-2 diabetes/insulin resistance accelerates brain aging by 4–7 years, and chronic inflammation (inflammaging) doubles the risk of substantial cognitive decline over the following two decades. Reducing branched-chain amino acid excess, insulin resistance, and inflammation therefore targets the exact upstream inputs the paper blames.
For the rapamycin data (rodent, not human): reported effects include a ~40% reduction in blood–brain barrier leakage, a 30–35% increase in hippocampal glucose uptake, prevention of the normal 25–35% loss of dendritic spines, a >60% reduction in senescent glia, and a doubling of neural progenitor proliferation within 7 days — all at plasma levels (3–8 ng/mL) already reached in human frailty trials.
Bottom line for a health-conscious reader: the lowest-risk actions are the AMPK-activating basics (exercise, avoiding insulin resistance, controlling inflammation). Rapamycin/rapalogs remain experimental for brain aging.
Context / Source
- Paywalled Paper: Unlocking the aging brain: mTORC1 as a convergent integrator for neurodegeneration and therapeutic intervention, 2026 Jun 15.
- Type: Review article.
- Authors / Institutions / Countries: Mokhtar Rejili (Imam Mohammad Ibn Saud Islamic University, Saudi Arabia); Hayder M. Al-kuraishy (Mustansiriyah University, Baghdad, Iraq); Mustafa M. Shokr (Sinai University–Arish, Egypt); Gaber El-saber Batiha (Damanhour University, Egypt).
- Journal: Biogerontology, 2026, Publisher: Springer Nature.
- Impact Evaluation: The impact score of this journal is 4.0, evaluated against a typical high-end range of 0–60+ for top general science, therefore this is a Medium impact journal.