Part 2: The Biohacker Analysis
Study Design Specifications
-
Type: Prospective Cohort Study (Level C).
-
Subjects: 208 healthy, non-obese (BMI < 30 kg/m²) human volunteers.
-
Note: Excluding obese individuals isolates IR as a variable independent of adiposity.
-
Methodology: Gold-Standard Insulin Suppression Test (IST). Subjects infused with somatostatin, insulin, and glucose to measure Steady-State Plasma Glucose (SSPG).
-
Follow-Up: 4–11 years (Mean: 6.3 ± 0.2 years).
Healthspan Analysis (The “Human Lifespan” Proxy)
-
Translational Gap: This is a human clinical study, not a murine lifespan experiment. Therefore, we measure “Event-Free Survival” rather than maximum lifespan.
-
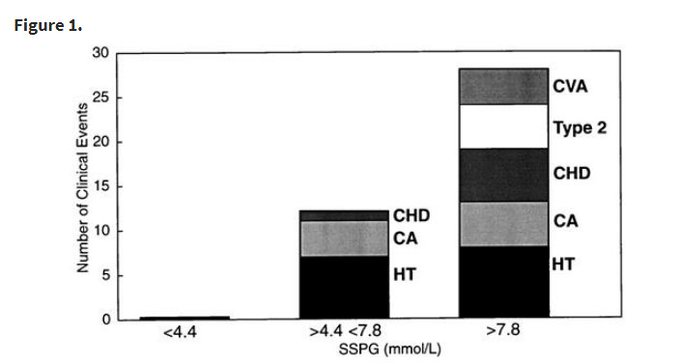
The “Zero-Event” Anomaly:
-
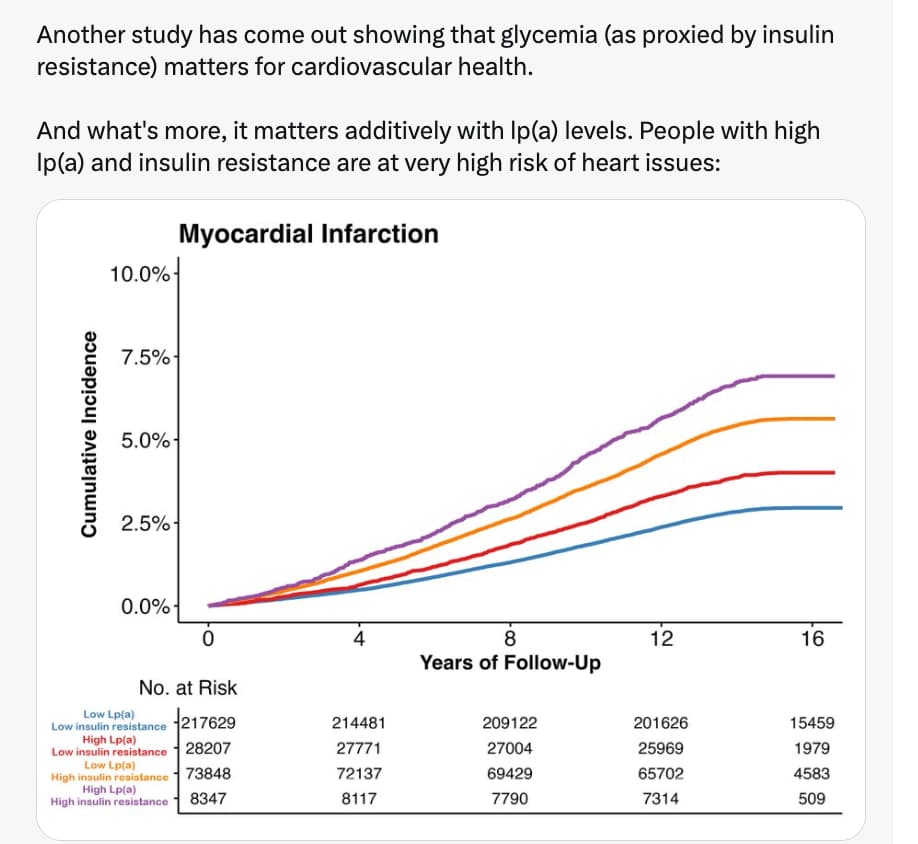
Insulin Sensitive Tertile (SSPG < 4.4 mmol/L): 0 clinical events. (100% Healthspan maintenance over duration).
-
Insulin Resistant Tertile (SSPG > 7.8 mmol/L): 28 clinical events (inc. Hypertension, Cancer, Stroke, CHD).
-
Risk Ratio: The risk difference is effectively infinite between the top and bottom tertiles during the observation window.
Mechanistic Deep Dive
-
The Hyperinsulinemia Toxicity Thesis: The study suggests that compensatory hyperinsulinemia (the body pumping out excess insulin to overcome resistance) is the primary driver of pathology, not just high glucose.
-
Oncology (IGF-1 Axis): The study observed 9 cancer cases, all in the insulin-resistant tertiles. High circulating insulin upregulates IGF-1 bioactivity and activates PI3K/Akt/mTOR pathways, promoting cellular proliferation and inhibiting apoptosis (a pro-tumorigenic state) Insulin Resistance and Cancer (2025).
-
Vascular Erosion: Insulin resistance abrogates NO-mediated vasodilation, leading to stiff, hypertensive arteries. The “Resistant” group had significantly higher blood pressure despite starting with normal vitals.
Critical Limitations
-
Sample Size: n=208 is relatively small for a prospective cohort.
-
Duration: ~6 years is insufficient to measure “Lifespan” extension, only mid-term “Healthspan.”
-
Causality: As an observational study, it proves prediction, not causation. However, the dose-response relationship (Tertile 1 vs 2 vs 3) strongly implies a biological gradient.
Part 3: Claims & Verification
Claim 1: Insulin Resistance predicts Coronary Heart Disease (CHD) and Stroke in non-diabetics.
-
Source Text: “Insulin resistance was an independent predictor of all clinical events… CHD + stroke.”
-
Verification: Supported. Large-scale meta-analyses confirm that HOMA-IR and fasting insulin are independent risk factors for CVD, even after adjusting for traditional risk factors.
-
Evidence Level: Level A (Meta-Analysis)
-
Live Search Validation: Insulin Resistance and CVD Risk (2012).
-
Consensus: High.
Claim 2: Insulin Resistance independently predicts Cancer.
-
Source Text: “Cancer was diagnosed in nine subjects… confined to the upper two tertiles.”
-
Verification: Supported (with nuance). While less established than CVD, modern data confirms that hyperinsulinemia is a carcinogen. Meta-analyses show specific links to breast, colorectal, and pancreatic cancers.
-
Evidence Level: Level A (Systematic Reviews)
-
Live Search Validation: Insulin resistance in cancer patients: Meta-analysis (2023).
-
Consensus: Medium-High. The link is robust, though the magnitude varies by cancer type.
Claim 3: Non-obese individuals can be metabolically obese (TOFI).
-
Source Text: “Baseline measurements… in 208 apparently healthy, nonobese (BMI < 30) individuals.”
-
Verification: Supported. This is the “Thin Outside, Fat Inside” (TOFI) phenotype. Visceral adipose tissue (VAT) drives IR regardless of subcutaneous fat.
-
Evidence Level: Level B/C.
-
Live Search Validation: Age, Obesity, and Insulin Sensitivity (2009).
Part 4: Actionable Intelligence
1. The Diagnostic Protocol (Finding your SSPG)
The study used the Insulin Suppression Test (IST), which is impractical for most biohackers (requires IV infusion of somatostatin). You must use proxies to estimate if you are in the “Zero-Event” tertile.
-
Gold Standard Proxy: Oral Glucose Tolerance Test (OGTT) with Insulin.
-
Action: Measure Glucose AND Insulin at 0, 30, 60, and 120 minutes after 75g glucose.
-
Target: Fasting Insulin < 5 uIU/mL; 1-hour Insulin < 30-40 uIU/mL.
-
Poor Man’s Proxy: HOMA-IR.
-
Formula: (Fasting Glucose mg/dL × Fasting Insulin uIU/mL) / 405.
-
Target: < 1.0 (Optimal); > 2.0 (Resistant).
-
Warning: HOMA-IR correlates with IST (r=0.8) but misses post-prandial dynamics HOMA vs Clamp Comparison (2014).
-
Lipid Proxy: TG/HDL Ratio.
-
Target: < 1.0 (mg/dL) or < 0.44 (mmol/L). High TG and Low HDL is the classic “Insulin Resistant Dyslipidemia” signature.
2. The Pharmacological Interventions
-
Metformin:
-
Mechanism: Activates AMPK, inhibits hepatic gluconeogenesis.
-
Dosing: 500mg - 1500mg ER (Titrate).
-
Safety: Check B12 levels annually. Avoid if eGFR < 30.
-
SGLT2 Inhibitors (e.g., Empagliflozin):
-
Mechanism: Excretes glucose via urine, lowers insulin demand independent of secretion.
-
Impact: Significant reduction in CVD and Renal events in non-diabetics.
-
Acarbose:
-
Mechanism: Alpha-glucosidase inhibitor. Blunts post-prandial glucose spikes, reducing the need for insulin surges.
-
Longevity: Proven to extend lifespan in mice (ITP study).
3. The Lifestyle Algorithm
-
Zone 2 Cardio: 150-180 mins/week. Increases mitochondrial density and GLUT4 translocation without spiking cortisol.
-
Muscle Mass: Skeletal muscle is the primary sink for glucose disposal (~80%). Hypertrophy training is non-negotiable for lowering SSPG.
-
Carbohydrate Tolerance: If you are in the upper tertile (Resistant), you effectively have “Carbohydrate Intolerance.” Restrict carbs to < 50g (Ketogenic) or use a CGM to keep post-prandial glucose < 110 mg/dL until sensitivity is restored.
Part 5: The Strategic FAQ
Q1: I have a normal A1c (5.1%). Does this mean I am insulin sensitive? A: No. A1c is a lagging indicator. You can maintain a normal A1c for years by hyper-secreting insulin (hyperinsulinemia) to force glucose into cells. This high insulin state drives cancer and heart disease risk before glucose fails. You must test Fasting Insulin.
Q2: What is the exact Fasting Insulin cutoff for the “Zero-Event” group? A: The study used SSPG, not fasting insulin. However, data correlates an SSPG < 4.4 mmol/L roughly to a Fasting Insulin of < 4–5 uIU/mL. If you are above 10 uIU/mL, you are likely in the high-risk tertile.
Q3: Can Rapamycin improve this metric? A: Paradox. Rapamycin inhibits mTORC1, which mimics fasting, but chronic high-dose Rapamycin can cause “benevolent pseudo-diabetes” (hepatic insulin resistance) by inhibiting mTORC2. Most longevity protocols use cyclical dosing (e.g., once weekly) to avoid this. Monitor HOMA-IR while on Rapamycin.
Q4: How does this relate to the “Lipid Hypothesis”? A: The study found that IR was an independent predictor, often stronger than LDL alone. In fact, IR drives small-dense LDL particles (Pattern B), which are more atherogenic. Correcting IR often fixes high Triglycerides and low HDL naturally.
Q5: Is “Post-Prandial” (after eating) or “Fasting” more important? A: The study used a steady-state infusion (simulating a fed state). Post-prandial clearance is the functional test of your system. A Fasting Glucose of 90 mg/dL is useless if you spike to 180 mg/dL and stay there for 3 hours after a meal.
Q6: Does this apply to women as well as men? A: Yes. The study included 110 females and 98 males. The tertile distribution and risk prediction held true across sexes.
Q7: I am “Skinny Fat.” Am I at risk? A: High Risk. The study specifically excluded obese people (BMI > 30), yet found massive pathology in the resistant group. These were likely “Skinny Fat” individuals with low muscle mass and visceral fat. Muscle is your metabolic armor.
Q8: Can I use a CGM to track SSPG? A: Not directly, but you can track Glycemic Variability (GV). High GV suggests poor insulin control. Aim for a standard deviation < 20 mg/dL.
Q9: What is the “HED” (Human Equivalent Dose) for lifestyle? A: There is no drug dose here, but the “Dose” of exercise required to move from Tertile 3 to Tertile 1 is estimated at 45 minutes of moderate activity daily combined with weight loss (if visceral fat is present).
Q10: Why haven’t I heard of the “Insulin Suppression Test” before? A: It is invasive, expensive, and risks hypoglycemia (requires continuous infusion of glucose and insulin). It is a research tool. HOMA-IR and LP-IR (Lipoprotein Insulin Resistance score) are the clinical replacements.
Incidence Rate of Insulin resistance predicts age-related diseases.
After 6 years, no one in lowest third got heart disease, stroke, cancer, hypertension, or diabetes.