

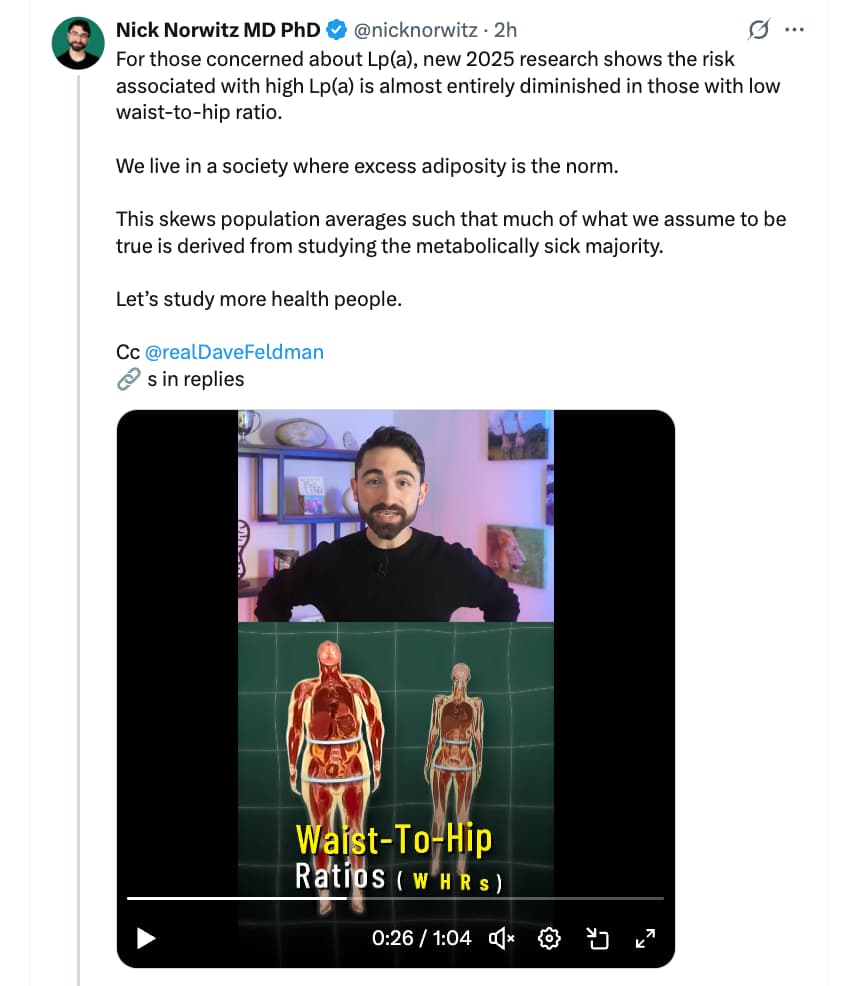
This is such great news! My Lp(a) was 40 and I was freaked out, even though CAC was 0.96 and I am metabolically healthy, BMI of 18.6. Have been on Repatha for a couple of years, and Ezetimibe too. Reduced Lp(a) to 27 and APOB also reduced. Did raise blood glucose some. Had been thinking of titrating off the Repatha and maybe also the Metformin. This latest information has really helped me move toward that decision. Suggestions / reactions would be welcomed!
3 Likes
My Lp(a) is 187. I am very thin (weigh 53 kg) so that is good news as my waist to him is good. Interesting aside- I had very high cholesterol past few years (subclinical hypo maybe caused it but I cant tolerate levo). A few months ago I started taking berberine one third of the dose suggested on the bottle (Natures Sunshine in Australia) and not every day. My latest blood test shows cholesterol in normal range- my doctor persobnally called me to ask what I was doing. Only other thing is we bought a water rowing machine and I do ten mins a day on that.
2 Likes
Thanks for sharing! This is super interesting. I clicked through to the paper and it does seem that the study was done in a reasonable way:
Methods: 4652 participants from the Multi-Ethnic Study of Atherosclerosis (MESA) were grouped as follows: Lp (a) <50 mg/dl and WHR <90th percentile(pct) (reference); Lp(a) <50 mg/dl and WHR ≥90th pct; Lp(a) ≥50 mg/dl and WHR <90th pct; and Lp(a) ≥50 mg/dl and WHR ≥90th pct.
Here, I wish they’d use a different cut-off. Here, 49mg/dl would be considered as “normal” when I would still call that elevated. I’d love to see the association in the <20mg/dl range, which I think is a majority of people.
Cox proportional hazard models assessed the relationship of Lp(a) and WHR with time to ASCVD events.
Results: Compared to the reference group, isolated elevated Lp(a) ≥50 mg/dl or WHR ≥90th pct were not significantly associated with risk of ASCVD (hazard ratio (HR), 1.15, 95 % confidence interval (CI): 0.94–1.39) and (HR, 1.14, 95 % CI: 0.92–1.41), respectively. In contrast, the combination of elevated Lp(a) ≥50 mg/dl and WHR ≥90th pct was associated with ASCVD risk (HR, 2.34, 95 % CI: 1.61–3.40).
Lp(a) ≥50 mg/dl was not significantly associated with ASCVD risk in the 1st and 2nd tertile of WHR (HR, 1.06, 95 % CI: 0.72–1.48and HR, 1.08, 95 % CI: 0.79–1.48, respectively). However, Lp(a) ≥50 mg/dl was significantly associated with ASCVD risk in the highest tertile of WHR (HR, 1.60, 95 % CI: 1.23–2.09). (Interaction p =0.01).
Body mass index (BMI) and Lp(a) combinations resulted in similar greater risks of ASCVD in the highest risk category (HR, 1.33, 95 % CI: 1.00–1.77), without a significant interaction (p =0.99).
The main limitation is that it’s a population based study looking at associations. It’s far from a guarantee that Lp(a) does no harm as long as you’re not fat. But I will take some encouragement that the signal for high Lp(a) alone is not strong enough to show up.
3 Likes
An update from WaPo:
So the cardiology community is closely watching a clinical trial seen as a bellwether for Lp(a) treatments.
The trial is studying pelacarsen, an experimental drug that stops the liver from producing the extra protein carried by Lp(a) that makes it especially risky. In an earlier trial, researchers showed the drug could reduce Lp(a) levels by up to 80 percent when injected weekly. Now the drug’s sponsor, Novartis, will be the first to reveal whether lowering Lp(a) levels also reduces cardiovascular events from patients who have heart disease.
Asked about pricing strategy on a November call with financial analysts, Novartis executives said that pelacarsen would initially be tailored to patients who’ve had early heart problems and a family history of disease, according to a transcript compiled by S&P Global Market Intelligence. “The family history is an emotional motivator for people to take action,” said Dianne Auclair Rocha, a senior vice president.
Though pelacarsen is the farthest along, other experimental drugs have shown they can lower Lp(a) even more sharply and for longer. Olpasiran, developed by Amgen, cut Lp(a) levels by up to 100 percent when taken every 12 weeks. Eli Lilly is studying lepodisiran, which works by a similar mechanism, to see if it reduces risk for patients who have not yet had a cardiac event — and it is also developing a pillfor lowering Lp(a).
“If these therapies show benefit, it would impact the lives of these individuals tremendously,” said Gissette Reyes-Soffer, an associate professor at Columbia University Irving Medical Center who advises companies targeting Lp(a). “You’re not going to have four stents put in,” she said, adding that preventing heart disease could save on health costs.
For now, there are few ways to lower Lp(a) levels. A class of cholesterol-lowering drugs has shown a modest effect, and an expensive blood-filtering procedure can also do so, though neither is approved by the Food and Drug Administration for that purpose. But some cardiologists bristle at physicians who decline to order tests for Lp(a) because there isn’t a drug that treats it.
“I think that’s crazy,” said Erin Michos, a professor of cardiology at the Johns Hopkins University School of Medicine. “I think Lp(a) is very actionable now,” she said, adding that physicians can take steps to lower all other treatable risks such as high cholesterol, blood pressure and weight. Michos has consulted for companies developing Lp(a) therapies.
Full article: A hidden risk to your heart has no treatment, but that could soon change (WaPo)
4 Likes

Just saw this on X from someone using Retatrutide. I am not sure how real it is. If this interests you, I would recommend doing more research on the topic:



Source: https://x.com/Oxandrolonely/status/2026717263424786500?s=20
7 Likes
Reading about all these potential health benefits is making me want to increase my dose and just force some extra calories in to offset it.
There doesn’t seem to be much out there on lp(a).
My unscientific search on perplexity:
I found only 2–3 anecdotal reports across the internet claiming retatrutide lowered Lp(a), out of thousands of retatrutide discussions.
Details on the reports
- One Reddit commenter in a Peter Attia thread explicitly says retatrutide “also reduced Lp(a), which is quite rare,” in the context of broader lipid benefits (no numbers given).
- An X (Twitter) post references “this guy lowered his Lp(a) by 70% on retatrutide,” linking to an unspecified example with 18 likes as of late February 2026.
And it appears there have been no studies other than this one for other glp’s that began in 2015:
Yes, proposing a trial reflects preliminary optimism from in vitro data showing GLP-1 agonists reduce Lp(a) synthesis in liver cells.
Why trials get proposed
Researchers at Vall d’Hebron University Hospital launched this study (NCT02501850, started 2015) specifically to test if liraglutide, exenatide, or lixisenatide lower Lp(a) in type 2 diabetes patients, building on their lab findings.
- The hypothesis explicitly states: “Treatment with GLP-1R agonists will lower the levels of Lp(a) in patients with DM-2.”
- It’s an observational setup: 20 patients newly prescribed a GLP-1 agonist vs. 20 on metformin/sulfonylurea, measuring Lp(a) at baseline and 2 months.
Status and results
The trial was listed as “recruiting” as of 2015 with no posted results by February 2026, so the hypothesis remains unconfirmed in humans.
- No published outcomes mean we lack evidence of Lp(a) reduction with these drugs, despite the belief it "might work."
- Broader GLP-1 meta-analyses (up to 2025) highlight cardiovascular benefits like reduced MACE and mortality but do not mention Lp(a) changes.
1 Like
Reta has had zero effect on my Lp(a), even at 6mg/week. I notice that the poster on X is posting on a forum (Oxandrolonely?). Oxandrolone is an anabolic steroid that does decrease Lp(a), so my guess is there’s a confounding variable involved here.
6 Likes
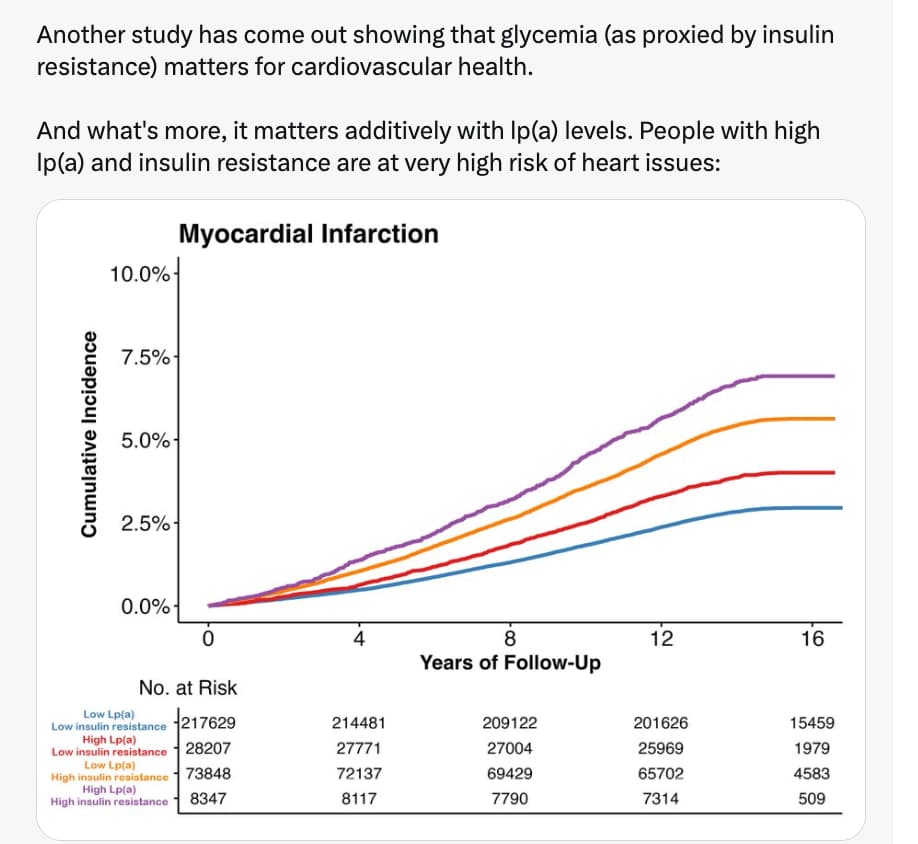
2 Likes
"Another study have come out showing glycemia…"?
Statins have been approved and studied to reduce risk in diabetics for a long time, who have high risk.
But you can’t get atherosclerosis with low apoB + Lp(a) for long as the genetic studies show. Ironically people believe insulin sensitivity does this.
1 Like
I just wanted to check this, so used this prompt:
and here is the result:
Atherosclerosis is fundamentally a disease of subendothelial lipoprotein retention and subsequent immune response. While apolipoprotein B (apoB) is the necessary causal agent for plaque formation, and Lipoprotein(a) [Lp(a)] is an independent causal risk factor, driving both to physiological minimums does not confer absolute immunity against atherogenesis.
Atherosclerosis can still progress with minimal circulating apoB and Lp(a) if endothelial permeability is compromised, vascular matrix proteins are modified, or the immune system is hyper-activated.
Here is the verified scientific and clinical consensus on lipid-independent atherogenic pathways.
Part 3: Claims & Verification
-
Claim 1: Residual Inflammatory Risk Drives Atherogenesis Independent of Lipids
- Mechanistic Summary: Even when atherogenic lipoproteins are aggressively lowered, persistent systemic inflammation—typically measured by high-sensitivity C-reactive protein (hsCRP)—drives endothelial dysfunction, upregulates adhesion molecules, and activates macrophages into foam cells.
- Evidence Level: Level A/B (Human Meta-analyses / Human RCTs).
- Supporting Evidence: The CANTOS trial demonstrated that inhibiting Interleukin-1β (a pro-inflammatory cytokine) significantly reduced major adverse cardiovascular events (MACE) without any reduction in apoB or LDL-C. Meta-analyses confirm that elevated baseline hsCRP predicts plaque progression regardless of statin or PCSK9 inhibitor intensity.
- External Verification: Residual inflammatory risk after contemporary lipid lowering therapy (2021)
-
Claim 2: Mechanical Stress and Hypertension Induce Endothelial Permeability
- Mechanistic Summary: Chronic hydrostatic pressure and disturbed shear stress physically damage the single-cell layer of the endothelium. This mechanotransduction downregulates protective nitric oxide (NO) synthase, forces cytoskeletal changes that widen intercellular gaps, and increases vascular permeability. This allows even trace amounts of circulating apoB to penetrate and become trapped in the intima.
- Evidence Level: Level C (Human Observational / Clinical Cohorts).
- Supporting Evidence: Clinical pathology confirms that structural alterations in resistance arteries and endothelial dysfunction often precede overt hypertension and are strictly correlated with future cardiovascular events.
- External Verification: Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications (2022)
-
Claim 3: Clonal Hematopoiesis of Indeterminate Potential (CHIP) Accelerates Plaque Formation
- Mechanistic Summary: As humans age, somatic mutations in hematopoietic stem cells (commonly in DNMT3A, TET2, ASXL1, and JAK2 genes) create mutant leukocyte clones. These clones drive a hyper-inflammatory macrophage phenotype that significantly increases atherothrombotic risk entirely independent of plasma lipid levels.
- Evidence Level: Level C (Human Cohort Studies) and Level D (Pre-clinical).
- Translational Gap: FLAGGED. The epidemiological association in humans is undeniable (CHIP roughly doubles the risk of coronary heart disease). However, the specific molecular pathways (e.g., TET2-driven chemokine overexpression) are heavily derived from Tet2 knockout hypercholesterolemic mice. How these specific macrophage pathways translate directly to human interventions remains unverified.
- External Verification: Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease (2017)
-
Claim 4: Advanced Glycation End-Products (AGEs) Modify the Vascular Matrix
- Mechanistic Summary: In states of impaired glucose tolerance, non-enzymatic glycation forms AGEs which cross-link with extracellular matrix proteins (like collagen), causing arterial stiffening.Binding to their receptor (RAGE) triggers intracellular oxidative stress and inflammatory cascades (NF-κB pathway), accelerating atherogenesis regardless of circulating apoB concentrations.
- Evidence Level: Level C (Human Cohorts) and Level D (Pre-clinical).
- Translational Gap: FLAGGED. Human studies show a strong correlation between AGE accumulation (via skin autofluorescence or serum) and atherosclerosis severity. However, claims that exogenous (dietary) AGEs directly cause plaque rupture rely heavily on high-fat, high-AGE feeding trials in genetic (db/db) murine models, which do not perfectly mirror human metabolic responses.
- External Verification: Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease (2020)
Knowledge Gaps & Actionable Insights
To achieve maximum longevity and cardiovascular risk mitigation, driving apoB and Lp(a) down is necessary but mathematically incomplete. Complete risk mitigation requires suppressing systemic inflammation (maintaining hsCRP well below 1.0 mg/L), maintaining strict normoglycemia to prevent matrix glycation, and optimizing blood pressure to preserve endothelial integrity.
The primary knowledge gap lies in the clinical management of CHIP. We currently lack human RCTs demonstrating whether targeted anti-inflammatory agents or specific lifestyle interventions can neutralize the cardiovascular risk conferred by these somatic mutations.
Would you like me to pull the clinical data on current pharmacological interventions (like colchicine or specific GLP-1 RAs) that are being used to target the residual inflammatory risk pathways?
1 Like
The key point is for long, which you missed:
But you can’t get atherosclerosis with low apoB + Lp(a) for long.
How does reducing inflammation independently of apoB+Lp(a) having an effect, refute my claim? Is it the same with smoking? It has nothing to do with it.
Find a single case of a heart attack following plaque rupture with total abetalipoproteinemia (LDL = 0).
1 Like
I’m not sure what you’re saying here. Are you saying you can’t get long-term atherosclerosis with low APOB and LPA? Or are you saying you need to have LP(a) for a long time to achieve atherosclerosis? I’m not sure what “for long” is referring to.
Yeah you can’t get atherosclerosis if you have low apoB + Lp(a) for a long time. At least the atherosclerosis we are familiar with today. Of course people have CVD with low apoB and Lp(a) in clinical trials which are short term. They’ve already had 40+ yrs of normal or high cumulative exposure.
You seem to have forgotten the Cohen 2006 study with PCSK9 loss of function, and that was only a slight decrease in LDL-C but over a long time.
3 Likes

I’m not sure that the above evidence hierarchy, which follows the conventional narrative, accurately reflects the underlying biological phenomena.
More precisely, I would put reliable human observational studies, like NANHES and Framingham, in place 2 together with human RCTs.
My reasoning is that big data + long duration + little accuracy is a complement to small data + small duration + high accuracy.
indisputably so, in big data = observational studies, the size of the sample makes inference to the whole population very rigorous. In other words, epistem ic uncertainty, related to the size of the sample compared to the size of the population, is almost nil. And we are almost sure not to miss very low or very high percentile data, that is, we are reasonably sure that the studied sample embraces the whole variability of the population.
1 Like