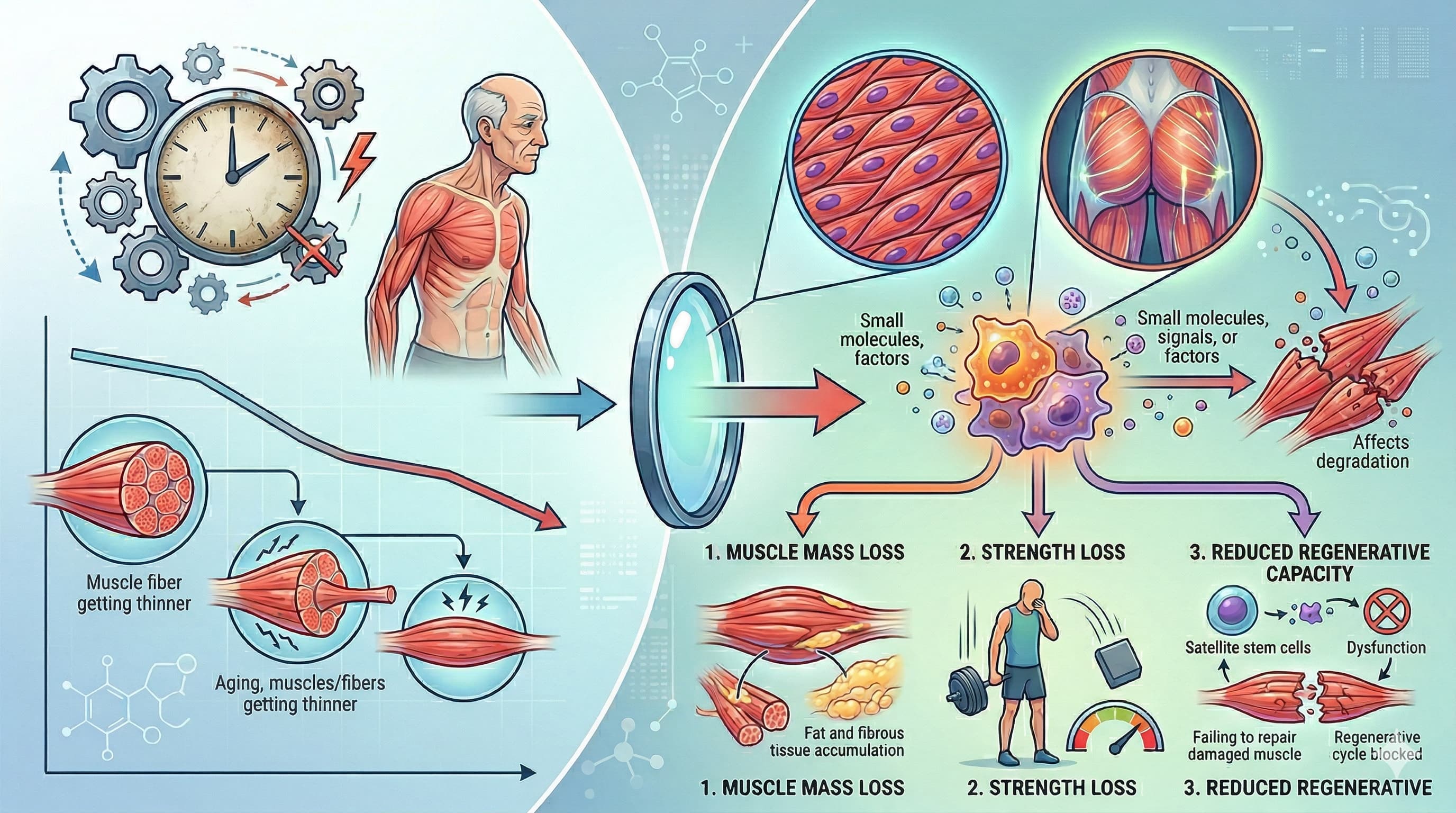
Skeletal muscle aging, clinically recognized as sarcopenia, has traditionally been viewed through the lens of passive “wear and tear.” However, recent evidence definitively shifts this paradigm toward an active, pathology-driving mechanism: cellular senescence. This review synthesizes mounting data demonstrating that the accumulation of senescent cells across various muscle compartments directly orchestrates the progressive loss of muscle mass, strength, and regenerative capacity.
Crucially, senescence in skeletal muscle is not confined to a single cell type. It actively infiltrates muscle stem cells (MuSCs), fibro-adipogenic progenitors (FAPs), immune cells, and even terminally differentiated, post-mitotic myofibers. Once senescent, these cells exhibit profound chromatin alterations, resistance to apoptosis, and the secretion of a toxic senescence-associated secretory phenotype (SASP). The SASP, a cocktail of pro-inflammatory cytokines, chemokines, and extracellular matrix-modifying enzymes, propagates chronic inflammation (“inflammaging”), disrupts niche homeostasis, and induces secondary senescence in neighboring healthy cells.
For example, senescent FAPs lose the ability to secrete WISP1—a vital regenerative signal—while simultaneously driving fibrotic tissue remodeling via transforming growth factor-β (TGF-β) secretion. Concurrently, senescent MuSCs suffer from impaired autophagy and profound regenerative deficits. Even post-mitotic myofibers, previously thought immune to classic replicative senescence, exhibit stress-induced premature senescence driven by p21CIP1 expression, further exacerbating tissue decline.
This mechanistic clarity has accelerated the development of senotherapeutics as a countermeasure. Senolytics (like the BCL-2/BCL-XL inhibitor ABT263 and CAR-T cells targeting uPAR) selectively induce apoptosis in senescent cells, thereby rejuvenating MuSC function in preclinical models. Senomorphics (such as the CCR5 antagonist Maraviroc and natural polyphenols) aim to suppress the SASP without eliminating the cells. While these pharmacological interventions present a formidable frontier against age-related frailty, clinical translation remains gated by target specificity, the complex heterogeneity of the senescent niche, and the potential off-target consequences of broad senescent cell clearance
Source
- Open Access Paper: Cellular Senescence in Skeletal Muscle Aging (Review article)
- Institution: Li Ka Shing Institute of Health Sciences, Chinese University of Hong Kong.
- Country: Hong Kong SAR, China.
- Journal: Endocrinology and Metabolism (EnM).
- Impact Evaluation: The impact score of this journal is 3.5, evaluated against a typical high-end range of 0–60+ for top general science, therefore this is a Medium impact journal.
Related Reading: HIV Drug (Maraviroc) Reverses Muscle Aging by purging “Zombie Cell” Signals