ACTIONABLE INTELLIGENCE (Translational Protocol)
Human Equivalent Dose (HED) — Showing the Math
FDA Guidance Used: “Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers” (FDA 2005), using body surface area (BSA) Km factor normalization.
Km Values (from FDA Table 1):
- Mouse Km = 3
- Human Km = 37 (based on 60 kg reference body weight)
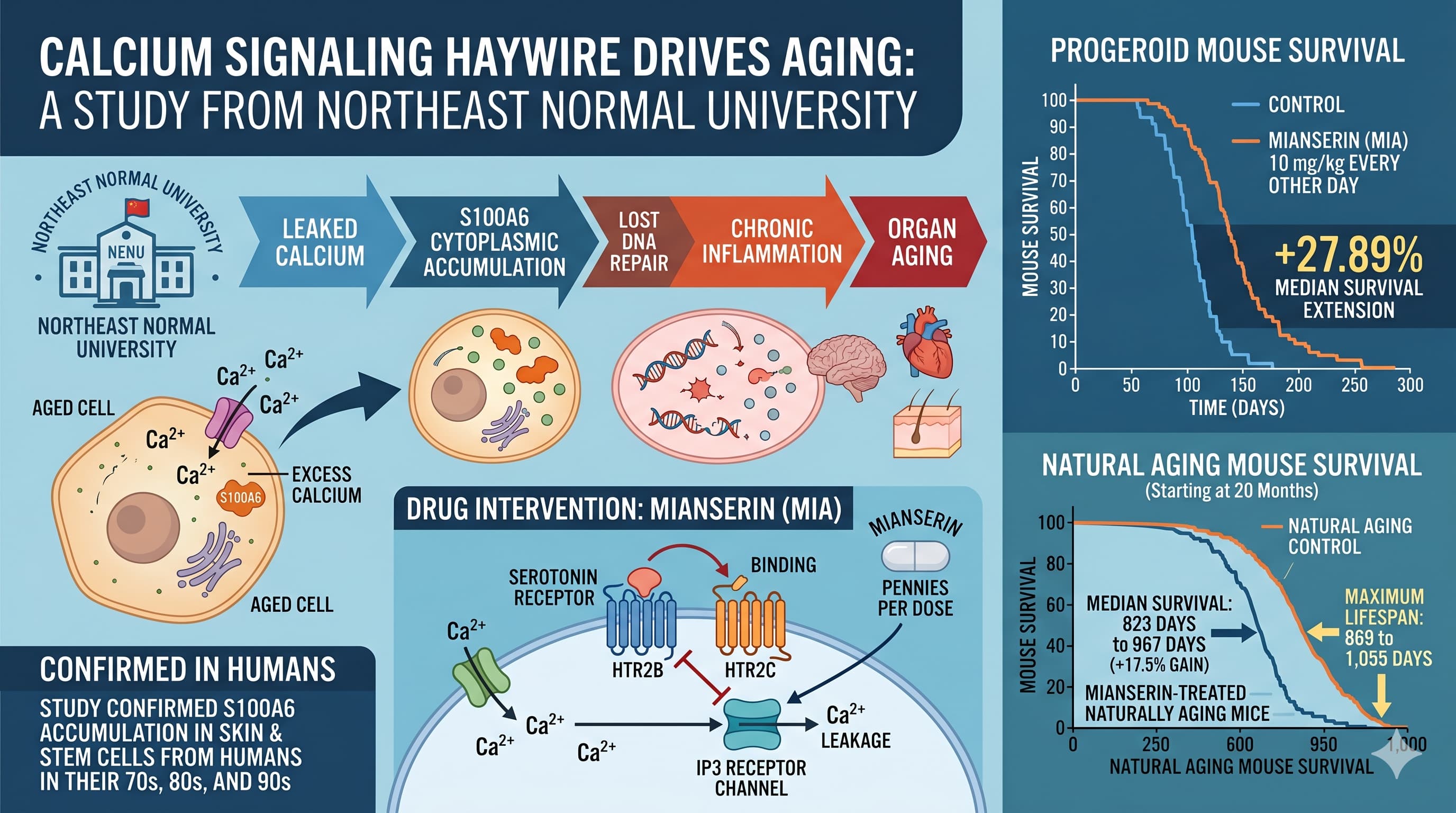
Study Dose: 10 mg/kg i.p., every other day, in mice.
Step 1 — Raw BSA-normalized HED:
HED (mg/kg) = Animal dose (mg/kg) x (Animal Km / Human Km)
HED = 10 mg/kg x (3 / 37) = 10 x 0.0811 = 0.81 mg/kg
For a 70 kg human: 0.81 mg/kg x 70 kg = 56.8 mg/day systemic equivalent
Step 2 — Route Correction (Critical Adjustment):
The mouse dose was intraperitoneal (i.p.), which gives near-complete systemic absorption (functionally equivalent to IV for this calculation). Mianserin’s oral bioavailability in humans is 20-30% (mean ~22-25%) due to extensive first-pass hepatic metabolism. To achieve the same systemic exposure orally, the dose must be corrected upward:
Oral dose needed = Systemic HED / Oral bioavailability
Oral dose = 56.8 mg / 0.25 = ~227 mg/day oral
Step 3 — Context Against Clinical Range:
The established therapeutic dose range for mianserin in depression is 30-90 mg/day (maximum 120 mg/day in some European protocols). The theoretical anti-aging HED of ~227 mg/day oral exceeds the clinical ceiling by approximately 1.9-7.6 times.
Interpretation: This is a significant red flag. The dose used in mice, when translated to a human oral equivalent via FDA BSA normalization with route correction, substantially exceeds the safe clinical dose range. There are two interpretive possibilities: (1) the effective plasma concentration in mice may be lower than the BSA calculation implies due to differences in volume of distribution and protein binding, or (2) the preclinical dose is genuinely supratherapeutic by human standards. Until plasma pharmacokinetic data from the mouse experiments are published, this dose mismatch must be treated as a serious translational barrier.
Mirtazapine as a proxy dose estimate: Mirtazapine (structurally related, same HTR2B/2C antagonism mechanism, FDA-approved) is used at 15-45 mg/day in humans. This is the most plausible clinical reference point for any anti-aging dosing strategy targeting this same receptor mechanism, though no direct anti-aging data exist for mirtazapine. [Confidence: Low — speculative extrapolation]
Pharmacokinetics and Pharmacodynamics
Oral Bioavailability: 20-30% (absolute bioavailability ~22% for solution, ~20% for tablets). Extensive first-pass hepatic metabolism is the primary limiter.
Tmax: 1.1-1.4 hours (oral). Rapid absorption.
Plasma Protein Binding: ~95%. High protein binding limits free fraction and extends half-life.
Elimination Half-life:
- Young adults: ~6-10 hours (some sources cite 6-39 hours depending on method)
- Elderly patients: mean 27 hours (range ~14-40 hours) due to reduced hepatic clearance
- The elderly half-life is clinically relevant because the target population for longevity use is older individuals, who will accumulate more drug and require dose adjustment.
Metabolism: Primarily hepatic. CYP2D6 is the major isoform (8-hydroxylation to form active 8-OH-mianserin; N-demethylation). CYP1A2 and CYP3A4 contribute to N-oxidation and ring hydroxylation. Active metabolites: 8-hydroxy-mianserin and desmethylmianserin — both retain pharmacological activity and contribute to total drug effect.
CYP2D6 Pharmacogenomics: CYP2D6 poor metabolizers (~5-10% of European populations, ~1-2% of East Asians) will have substantially higher mianserin plasma levels and longer effective half-lives, compounding the dosing complexity.
PD Target Engagement: Mianserin’s mechanism in this study is HTR2B/2C antagonism → IP3R suppression → reduced ER Ca2+ release. The drug-receptor binding is competitive and reversible. Given the every-other-day dosing used in mice and the short half-life in young animals, receptor occupancy would have been intermittent. In elderly humans with longer half-lives (~27 hours), less frequent dosing might achieve comparable receptor occupancy, which is relevant for optimizing a safety-favorable dose schedule.
Safety and Toxicity
Acute Toxicity:
- LD50 (oral, rat): 780 mg/kg. Therapeutic index relative to the theoretical human oral HED (~227 mg/day, ~3.2 mg/kg for 70 kg) is approximately 240:1 on a mg/kg basis. This appears acceptable in isolation but must be weighted against the chronic toxicity data below.
NOAEL (Repeat-Dose): Safety Data Absent. No published NOAEL from a formal repeat-dose GLP toxicology study was identified in a literature search. The NOAEL for long-term dosing in the anti-aging context (years of continuous use) is therefore unknown.
Hematological Toxicity — CRITICAL: Bone marrow depression, specifically agranulocytosis and granulocytopenia, is the most serious adverse effect. Reported incidence: approximately 1:2,000 to 1:4,000 exposures in clinical use. Onset is most common at 4-6 weeks of treatment. In elderly patients, the incidence may be higher. Standard monitoring protocol: complete blood count (CBC) every 4 weeks for the first 3 months of treatment, with immediate discontinuation and CBC if infection symptoms develop (fever, sore throat, stomatitis). For a longevity use case implying years of continuous treatment, the cumulative agranulocytosis risk and the monitoring burden are non-trivial considerations.
Cardiac Safety:
- No clinically significant QTc prolongation at therapeutic doses. No anticholinergic cardiac effects (advantage over TCAs).
- Overdose can produce sedation, coma, hypotension/hypertension, tachycardia, and QT prolongation.
- No significant cardiac valvulopathy risk (MIA is an HTR2B antagonist, not an agonist — HTR2B agonism, as with fenfluramine, causes valve disease; antagonism does not share this liability).
CNS Effects:
- Sedation via H1 (histamine) antagonism — dose-dependent. A sedating side effect in an elderly longevity user could increase fall risk, a critical safety consideration in geriatric populations.
- Weight gain via H1 + 5-HT2C antagonism — potentially problematic for metabolic health if used long-term.
- Orthostatic hypotension via alpha-2 adrenergic blockade — fall risk in elderly.
- No anticholinergic burden (advantageous).
- No significant dependence or withdrawal syndrome reported.
Overdose Profile: Mianserin is substantially safer in overdose than tricyclic antidepressants. Lethal cardiac arrhythmias do not occur at overdose doses that produce coma in TCAs.
CYP450 Drug Interaction Risk:
- CYP2D6 inhibitors (fluoxetine, paroxetine, bupropion) will raise mianserin plasma levels — potentially increasing sedation, weight gain, and agranulocytosis risk.
- CYP1A2 inducers (smoking, rifampin) may reduce mianserin levels.
- CYP3A4 interactions: weak — not a major concern at standard doses.
- Mianserin does not appear to be a significant inhibitor or inducer of CYP450 enzymes at therapeutic concentrations.
Biomarker Verification: Target Engagement Markers
To confirm that a given dose of mianserin is engaging the Ca2+/S100A6/cGAS-STING pathway in humans, the following measurable endpoints are recommended based on the paper’s mechanistic framework:
Primary Target Engagement (most direct):
- Intracellular Ca2+ in peripheral blood mononuclear cells (PBMCs): measurable by flow cytometry using Fluo-4 AM dye. A reduction in cytoplasmic Ca2+ in PBMCs post-treatment would confirm on-target receptor-level action.
- S100A6 plasma levels: ELISA-measurable. An aging biomarker that should decrease with effective treatment.
- S100A6 cytoplasmic vs. nuclear localization: requires skin punch biopsy (3-4mm) + immunofluorescence. More invasive but most mechanistically direct.
Downstream Pathway Markers (confirmatory):
- PARP1 protein levels in PBMCs: Western blot or ELISA. Should increase with effective treatment.
- SASP cytokine panel in plasma: IL-6, IL-8, CXCL10, TNF-alpha, IL-1beta. These are standard commercial multiplex panels (e.g., Luminex, Olink).
- p-STING and p-NF-kB p65 in PBMCs: phospho-flow cytometry; specialized but available at major academic centers.
Surrogate Pharmacodynamic Marker (easiest, least specific):
- Serum serotonin (5-HT): The paper showed MIA increases serum 5-HT in progeroid mice. Serum 5-HT is easily measurable by standard clinical ELISA and could serve as a simple PK/PD surrogate for HTR2B/2C receptor occupancy.
Systemic Aging Biomarkers (least specific but broadest relevance):
- hs-CRP and IL-6 (systemic inflammation)
- Epigenetic clocks (DunedinPACE, GrimAge, PhenoAge) — would require 6-12 months minimum to show meaningful change
- p16-INK4a (CDKN2A) expression in T-cells — an established senescence burden marker
Feasibility and ROI
Sourcing:
Mianserin is NOT FDA-approved and is not legally available by US prescription. In the United States, access options are:
- Research chemical suppliers (Sigma-Aldrich, Cayman Chemical, MedChemExpress, Tocris): Sold as HCl salt for research purposes only. Typical price: ~$30-80 for 250mg-1g powder. No pharmaceutical-grade manufacturing, no dosage form standardization, no clinical quality control. Not recommended for human self-experimentation.
- International pharmacy (UK, EU, Asia): Mianserin is available by prescription in the UK (NHS), Europe, and parts of Asia. Prices in the UK are approximately GBP 5-15 for 28 tablets at 10-30 mg. International mail-order is a legal gray area under US law.
- Mirtazapine as a proxy: Mirtazapine (Remeron) shares the same HTR2B/2C antagonism mechanism, is FDA-approved in the US, and is available as a cheap generic (~$5-15/month for 15-30mg tablets). It has NOT been tested for anti-aging effects, but is the most plausible accessible analog targeting the same upstream pathway. This substitution is entirely unvalidated and speculative.
Cost vs. Effect Estimate:
Assuming access to pharmaceutical-grade mianserin at UK NHS pricing (~GBP 10/28 tablets at 30 mg), and a target dose of ~30-60 mg/day (below the theoretical HED but within the therapeutic range, acknowledging the HED dose gap):
Monthly cost for mianserin (30 mg/day): approximately USD 15-30/month at international pharmacy prices.
Marginal gain estimate: The 17.5% median lifespan extension in mice, discounted heavily for (1) single unreplicated study, (2) single sex, (3) borderline short-lived controls, (4) route/dose uncertainty, and (5) no human data — yields a very wide confidence interval around any human benefit projection. The probability that mianserin extends human lifespan is, at this stage, speculative. The drug cost is low; the unknowns are high.
Bottom line on ROI: Cost is negligible relative to most longevity interventions. The risk:benefit ratio is currently unfavorable for clinical self-experimentation due to (1) agranulocytosis risk requiring active hematological monitoring, (2) sedation and weight gain as counterproductive metabolic side effects, (3) the unresolved HED dose gap, and (4) complete absence of human pharmacodynamic or efficacy data. This calculus may change with replication data or a human PD pilot study.
What are the interactions between mianserin and the most common longevity stack drugs (rapamycin, metformin, SGLT2 inhibitors, acarbose, 17-alpha estradiol, PDE5 inhibitors)?
Answer, by compound:
Rapamycin: Rapamycin is a CYP3A4 substrate and P-glycoprotein substrate. Mianserin is metabolized primarily by CYP2D6 with minor CYP3A4 involvement. No clinically significant PK interaction is expected. Mechanistically, rapamycin suppresses mTORC1 (inhibiting anabolic signaling) while mianserin targets Ca2+/cGAS-STING (suppressing senescence-associated inflammation). These pathways are distinct and potentially complementary; additive anti-SASP effects are biologically plausible. No combination data exist. Additive immunosuppression is theoretically possible but unlikely at longevity doses of rapamycin.
Metformin: Metformin is renally excreted and not CYP-metabolized — no PK interaction. Metformin modestly reduces circulating serotonin levels in some studies, which could theoretically affect the same 5-HT signaling axis MIA acts on, but this has not been studied. PD overlap: both have anti-inflammatory effects through different mechanisms; additive reduction in SASP is plausible. No meaningful clinical interaction expected.
SGLT2 inhibitors (empagliflozin, dapagliflozin): Metabolized by UGT glucuronidation — minimal CYP involvement, no PK interaction with mianserin. SGLT2 inhibitors reduce intracellular sodium and secondarily affect Na+/Ca2+ exchanger activity in cardiomyocytes, which modestly reduces mitochondrial and cytoplasmic Ca2+ overload. This is a mechanistic overlap with MIA’s Ca2±lowering effect, potentially additive for cardiac protection. Clinically, the combination is not contraindicated.
Acarbose: Gut-restricted, negligible systemic absorption, no PK or PD interaction with mianserin.
17-alpha estradiol: CYP1A2 and CYP3A4 substrate. Mild potential competition with mianserin for CYP1A2, which could marginally elevate mianserin levels, but this is unlikely to be clinically significant at the doses used in longevity protocols. No data on this combination exist.
PDE5 inhibitors (sildenafil, tadalafil): CYP3A4 substrates. Mianserin is not a significant CYP3A4 inhibitor, so no major PK interaction. Both drug classes produce vasodilation — mianserin via alpha-2 adrenergic blockade (increasing norepinephrine release paradoxically, but also peripheral vasodilation), PDE5 inhibitors via cGMP/NO. Additive hypotension and orthostatic hypotension is the main clinical risk, particularly relevant in older adults where fall risk is a priority concern. This combination should be used cautiously with blood pressure monitoring.
You treated mice starting at 20 months — roughly equivalent to a 65-year-old human. Is there evidence for or against a critical window effect? Would starting MIA earlier (e.g., at 12 months, equivalent to middle age) produce greater or smaller benefits?
Answer: The paper tested only one age of treatment initiation (20 months) in natural aging mice. No cohort starting treatment at 12 months (middle age) or earlier was included. Evidence from other longevity interventions is mixed: rapamycin shows greater lifespan extension when started earlier (9 months vs. 20 months in most ITP studies), while some senolytic interventions show equivalence across start ages. For MIA specifically, its mechanism targets a pathway (Ca2+ dysregulation → S100A6 accumulation) that develops progressively with age, suggesting earlier intervention might prevent more accumulated damage. On the other hand, the Ca2+ dysregulation and HTR2B/2C upregulation in HGPS and aging are established features of already-aged cells, so intervention at the time of established dysregulation makes mechanistic sense. The answer is genuinely unknown, and the critical window question has direct practical relevance for a human longevity protocol. [Confidence: Low — pure speculation without data]