Looking deeper at the possible therapeutics for this neurodegeneration issue, it doesn’t look very good. MitoQ has failed in all the clinical trials its been tried in related to neurodegeneration (Parkinson’s etc.) but may still show results in terms of prevention. Ceftriaxone is IV delivery, which is impractical, and it can’t be used on an ongoing (daily) basis. Lions mane mushrooms “might” provide some benefits, but still unproven. The SS-31 peptide has some potential, but sourcing high quality at reasonable cost is an issue.
Executive Summary: The Translational Reality of MitoQ in Neurodegeneration
Despite possessing a mountain of highly successful pre-clinical data in rodent models of Alzheimer’s, ALS, traumatic brain injury, and Huntington’s disease, MitoQ’s human clinical trial record for neurodegenerative disease is exceptionally sparse and currently stands at a 100% failure rate for disease modification.
The stark reality of mitochondrial-targeted antioxidants is a massive translational gap. While the molecule successfully concentrates in the mitochondria of isolated cells and rodent brains, scaling the dose to achieve therapeutic central nervous system (CNS) penetrance in living humans frequently triggers dose-limiting gastrointestinal toxicity before neuroprotection can be achieved. Furthermore, quenching reactive oxygen species (ROS) appears insufficient to halt the cascading structural collapse of advanced human proteopathies (like Parkinson’s).
Active / Ongoing Clinical Trials
Because MitoQ failed to alter the trajectory of established Parkinson’s, current clinical research has pivoted away from “late-stage disease modification” and toward early-stage vascular dementia, Mild Cognitive Impairment (MCI), and brain hypoperfusion.
3. Mild Cognitive Impairment (MCI) & Cerebrovascular Blood Flow
4. The Mito-Frail Trial: Cognitive and Physical Dysfunction
The Biohacker & Longevity Takeaway
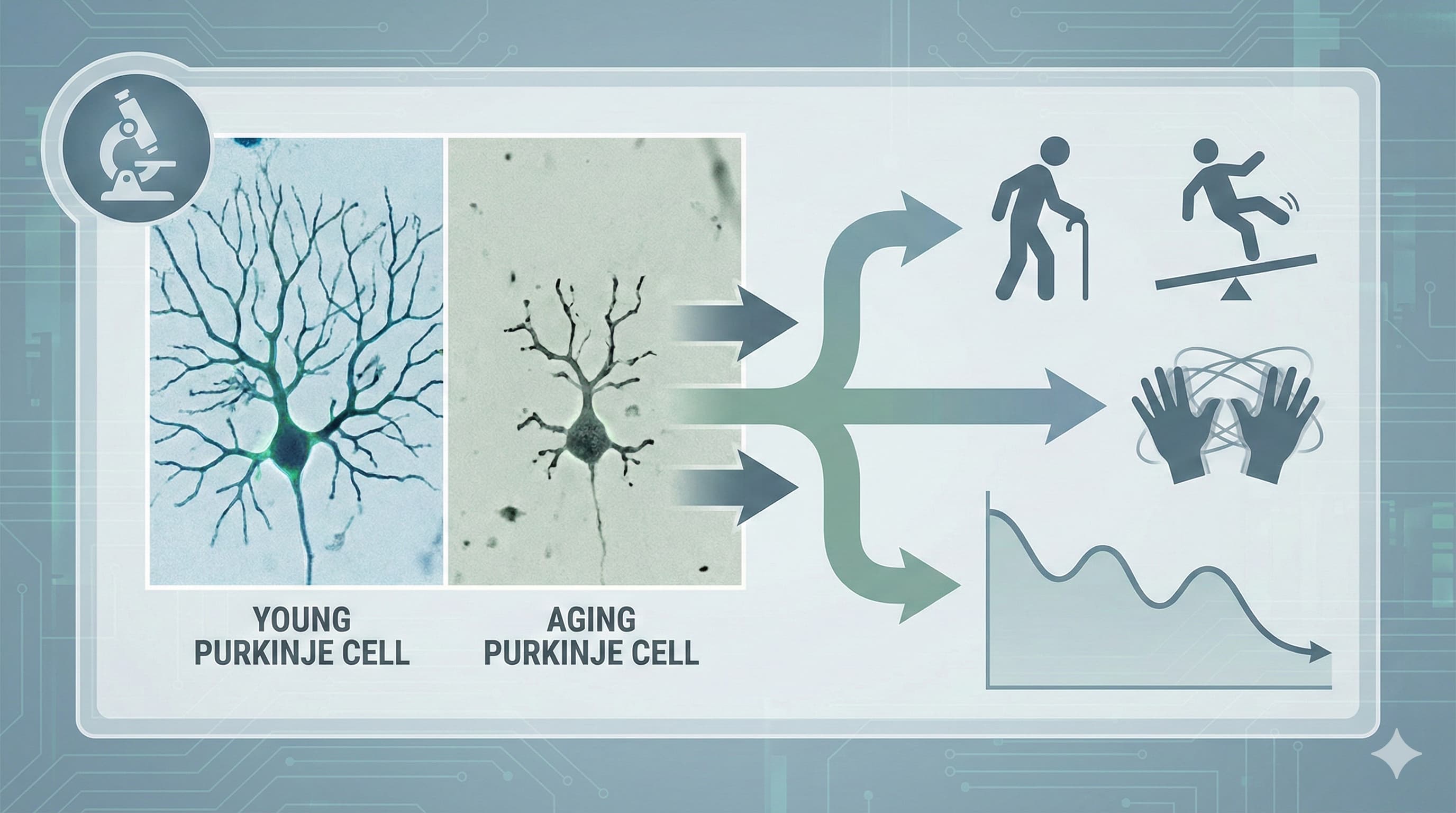
If your goal is structural neuroprotection (e.g., preserving Purkinje cells or dopaminergic neurons), human clinical data does not currently support the use of MitoQ. The failure of the PROTECT trial highlights two critical pharmacological hurdles:
-
The Blood-Brain Barrier (BBB) Bottleneck: Achieving therapeutic concentrations of MitoQ inside human brain mitochondria requires pushing systemic oral doses to levels that trigger severe GI toxicity (80+ mg). The standard commercial dose of 10–20 mg/day is profoundly unlikely to reach the CNS in concentrations high enough to halt neurodegeneration.
-
Too Little, Too Late: In neurodegenerative pathologies, by the time clinical symptoms manifest, the structural network collapse is too severe to be reversed simply by capping mitochondrial reactive oxygen species.
Currently, MitoQ’s validated human clinical utility remains strictly confined to peripheral vascular health (improving endothelial flow-mediated dilation in older adults). It should not be prescribed or relied upon as a primary neuroprotective agent.
The Ceftriaxone Translational Protocol:
-
The Clinical Precedent: Ceftriaxone is an FDA-approved, third-generation cephalosporin antibiotic. Its neuroprotective mechanism is completely independent of its antimicrobial properties; it transcriptionally upregulates the GLT-1 (EAAT2) glutamate transporter on astrocytes, enhancing the clearance of excitatory glutamate from the synaptic cleft and preventing calcium-induced excitotoxicity in neurons.
Pharmacokinetics (PK/PD):
-
Bioavailability: Zero to <1% orally. The drug is completely degraded in the GI tract. It strictly requires intravenous (IV) or intramuscular (IM) administration.
-
Phase I / Clinical Safety: Extremely well characterized as a short-term antibiotic (Rocephin). However, chronic daily administration is highly problematic. The primary long-term risk is biliary pseudolithiasis (gallbladder sludging), caused by the precipitation of ceftriaxone-calcium salts.
-
NOAEL / LD50: Acute toxicity is exceptionally low. The intravenous LD50 in mice is ~2,000 mg/kg. Long-term tox studies in non-human primates established a NOAEL of approximately 100 mg/kg/day due to gastrointestinal and biliary complications.
The Hericium erinaceus (Lion’s Mane)Translational Protocol:
-
The Clinical Mechanism: Hericium erinaceus (Lion’s Mane) acts as a neurotrophic stimulator. Its primary bioactive compounds—specifically erinacines—are small-molecule cyathane diterpenoids capable of crossing the blood-brain barrier (BBB). Once in the central nervous system, they stimulate the synthesis of Nerve Growth Factor (NGF) and Brain-Derived Neurotrophic Factor (BDNF), protecting neurons against endoplasmic reticulum stress, excitotoxicity, and apoptosis.
-
Human Equivalent Dose (HED): Pre-clinical models evaluating age-related cognitive decline (e.g., SAMP8 mice) and neurodegeneration utilize Erinacine A-enriched mycelium doses ranging from 108 mg/kg to 300 mg/kg per day.
-
Human Actionable Target: Using a moderate efficacy dose of 215 mg/kg: Mouse Dose 215 mg/kg × (Mouse Km 3 / Human Km 37) = 17.43 mg/kg. For a 70 kg human, this translates to ~1,220 mg/day (1.2 grams) of standardized mycelium extract.
Pharmacokinetics (PK/PD) of Erinacine A:
-
Bioavailability: ~24.39% orally. While modest, it is highly lipophilic and sufficient to achieve CNS penetrance.
-
Half-life: Approximately 7.3 hours in mammalian models. Following oral administration, Erinacine A is detectable in cerebrospinal fluid (CSF) within 15 minutes and reaches peak concentrations in brain tissue between 1 and 8 hours.
-
Clearance: Rapid hepatic processing. Over 75% of the compound is metabolized into secondary metabolites (like Erinacine B) within 60 minutes via the liver.
Feasibility & ROI:
-
Sourcing: Widely available over-the-counter (OTC) globally.
-
Cost vs. Effect: ~$20–$50/month. The ROI for generalized neuroprotection and cognitive maintenance is high, provided the user sources the correct formulation (mycelium, not fruiting body). The ROI for reversing acute cerebellar ataxia or severe motor decline is zero.
The Strategic FAQ
1. Does standard Lion’s Mane powder from the grocery store protect Purkinje cells? Answer: Highly unlikely. The bioactive compounds proven to cross the BBB and stimulate NGF are erinacines, which are almost entirely restricted to the mushroom’s mycelium (the root-like structure). Standard culinary powders and cheap supplements use the fruiting body (the visible mushroom cap), which primarily contains hericenones—compounds that largely fail to stimulate clinically significant neurogenesis.
2. If Erinacine A promotes NGF, does it actually rescue Purkinje cell firing rates as discussed in the McGill paper? Answer: Unknown. Pre-clinical data proves Lion’s Mane prevents structural Purkinje cell death (apoptosis) and stimulates neurite outgrowth in the presence of toxins. However, there is absolutely no ex vivo electrophysiological data proving it restores the intrinsic, high-frequency electrical pacemaking of aged Purkinje cells. It protects the hardware, but we do not know if it tunes the electrical signal.
3. What is the primary physiological bottleneck for this supplement? Answer: First-pass liver metabolism. While Erinacine A has an absolute oral bioavailability of ~24%, LC-MS/MS data shows that roughly 75% of the circulating compound is aggressively metabolized into secondary metabolites (like Erinacine B) within the first 60 minutes of ingestion.
4. Can stimulating Nerve Growth Factor (NGF) cause neuropathic pain? Answer: Theoretically, yes; clinically, no. Elevated NGF in peripheral tissues is highly correlated with hyperalgesia (increased pain sensitivity) and is a target for anti-pain biologics. However, systemic oral dosing of Lion’s Mane has never been reliably linked to inducing nerve pain in human cohorts, likely because its effects are modulatory rather than artificially flooding the system.
5. Is there definitive human clinical data for severe neurodegeneration? Answer: No. Small-scale, double-blind human trials show mild improvements in spatial memory and cognitive function in older adults with Mild Cognitive Impairment (MCI). There are no massive Phase III trials proving it alters the disease trajectory of hard neurological pathologies like Spinocerebellar Ataxia, ALS, or advanced Alzheimer’s.
6. Does Lion’s Mane carry bleeding risks? Answer: Mildly. The mushroom possesses mild anti-platelet aggregation properties. While entirely safe for a healthy biohacker, it introduces an additive bleeding risk if combined with prescription anticoagulants (e.g., warfarin, apixaban, clopidogrel).
7. Is this a viable replacement for targeted pharmacological therapies like 4-Aminopyridine for motor decline? Answer: Absolutely not. 4-AP is a direct voltage-gated potassium channel blocker that immediately lowers the action potential threshold, forcing neuronal firing within hours. Lion’s Mane is a slow-acting neurotrophic agent that requires weeks to alter gene transcription and NGF levels. It offers zero immediate symptomatic relief for motor coordination.
8. Why do some clinical trials and user anecdotes show zero effect? Answer: Lack of standardization in the supplement industry. If a product is not explicitly standardized to a guaranteed percentage of Erinacine A (e.g., 5 mg/g) and utilizes unfermented fruiting bodies rather than liquid-cultured mycelium, the user is consuming inert dietary fiber.