Actionable Intelligence
The Translational Protocol: Urolithin A (UA)
We will focus this protocol on Urolithin A, which the authors highlight as a primary clinical intervention for mitochondrial quality control and mitigating upstream drivers of inflammation.
-
Human Equivalent Dose (HED): Preclinical mouse models frequently demonstrate muscle and longevity benefits at an oral dose of 50 mg/kg/day.
-
Math (FDA BSA Normalization): 50 mg/kg×(3/37)=4.05 mg/kg. For a standard 70 kg human, the theoretical HED is 283.5 mg/day.
-
Correction for human metabolism: Current human clinical trials successfully utilize 500 mg to 1,000 mg daily [ClinicalTrials.gov]. This indicates that standard allometric scaling significantly underestimates the effective human dose, largely due to extensive human Phase II metabolism (glucuronidation and sulfation) in the enterocytes and liver [PubChem].
-
Pharmacokinetics (PK/PD): * Half-life: ~17–22 hours.
-
Bioavailability: Reliance on dietary precursors (ellagitannins from pomegranates or walnuts) is highly inefficient; only 30-40% of humans possess the necessary gut microbiota to convert these to UA. Direct oral supplementation (e.g., Mitopure) circumvents this bottleneck, increasing plasma UA levels roughly 6-fold compared to diet. UA is lipophilic and successfully penetrates the blood-brain barrier (BBB) [ADDF Report].
-
Safety & Toxicity:
-
NOAEL: Extremely high. >3,451 mg/kg bw/day in male rats (the highest dose tested in a 90-day oral study) [PubMed].
-
LD50: Not definitively reached in standard acute toxicity assays; the FDA has granted it Generally Recognized as Safe (GRAS) status.
-
Phase I safety profile: Clinically proven to be well-tolerated up to 1,000 mg/day for 4 months in older adults with no serious adverse events reported.
-
CYP450 / Organ Signals: UA may interact with CYP3A4. In D-galactose-induced aging models, it exerts active hepatoprotective and nephroprotective effects by reducing malondialdehyde (MDA) [RSC Advances]. Critical Caveat: UA must be approached with extreme caution in immunosuppressed or renal transplant patients, as its effects on calcineurin inhibitor metabolism and transplanted organ physiology are completely unverified.
Biomarker Verification
Target engagement can be verified via specific downstream blood panels. Successful intervention should yield:
-
Systemic Markers: Reduced plasma ceramides (a validated CVD risk predictor) and reduced C-reactive protein (CRP).
-
Metabolic Markers: Favorable modulation of plasma acylcarnitines, indicating improved mitochondrial turnover.
-
Cellular Markers: Advanced flow cytometry will show increased mitochondrial mass and reduced inflammatory cytokine expression (IL-6, TNF-α) specifically in CD3+/CD8+ T-cells [ASCO Abstract]. Muscle biopsies confirm the upregulation of mitophagy gene signatures.
Feasibility & ROI
-
Sourcing: Broadly available as an over-the-counter dietary supplement, most notably under the patented Mitopure formulation (Amazentis/Timeline Nutrition).
-
Cost vs. Effect: Approximately $100–$150/month for the clinical dose of 500 mg–1,000 mg/day. The ROI is high for adults (50+) presenting with declining muscular endurance, elevated baseline CRP, or metabolic syndrome. The marginal gain for healthy, highly active individuals under 35 remains unproven and is likely low.
The Strategic FAQ
1. Are the elevated levels of circulating SASP markers (like GDF15) truly causal in atherosclerosis, or are they purely compensatory bystanders to the real driver (hypoxia)? Based on the paper, they initially function as a compensatory resilience mechanism to alert surrounding tissues of metabolic or mitochondrial stress. However, chronic, unresolved secretion becomes maladaptive, actively degrading the extracellular matrix and compromising plaque caps, making them secondary drivers of the disease.
2. How do we accurately measure “chronic microvascular hypoperfusion” in asymptomatic patients before irreversible fibrosis occurs? This remains a critical clinical blind spot. The authors admit that clinical signs only appear in severe states, and current diagnostic tools—such as circulating lactate or sidestream dark-field imaging—are either biologically insensitive to mild hypoxia or not user-friendly in standard clinical practice.
3. If we successfully clear senescent endothelial cells using senolytics like Dasatinib + Quercetin, how does the vascular tissue regenerate without exhausting the local stem cell niche? The paper does not resolve this paradox. It notes that senescent endothelial cells impair regenerative capacity, but it is a known biological trade-off that aggressively clearing senescent cells requires a competent progenitor pool to replace them, which is itself diminished by age-related entropy.
4. Urolithin A induces mitophagy, but could chronic, high-dose administration trigger excessive mitochondrial depletion in energy-demanding tissues like the heart? Phase I trials and preclinical data indicate UA acts as an optimizer, clearing defective mitochondria and stimulating biogenesis without causing pathological depletion. However, robust data beyond 4 months of continuous human use is currently absent.
5. Is the accumulation of oxidized LDL the initiator of endothelial senescence, or does pre-existing, stochastically driven endothelial senescence allow for the accumulation of oxidized LDL? It is a pathological feedback loop. Initial stressors like ROS and turbulent blood flow promote LDL entry and oxidation in the subendothelial space. This insult pushes endothelial cells into senescence, upregulating adhesion molecules (ICAM1) that recruit macrophages to ingest oxLDL, turning them into foam cells.

6. Given the Unfolded Protein Response (UPR) and Integrated Stress Response (ISR) are essential for handling acute proteostatic stress, wouldn’t chronically suppressing them to lower inflammation increase the risk of protein aggregation diseases? Absolutely. The authors emphasize that these stress response pathways are protective. Longevity interventions must aim to resolve the upstream damage (e.g., hypoxia or nutrient stress) rather than directly silencing the UPR/ISR, which could lead to fatal cellular dysfunction.
7. Your proteomic analysis highlights non-cardiovascular tissues (kidney, liver) as major sources of CVD biomarkers. Does this mean targeting vascular tissue alone is a fundamentally flawed approach to CVD? Yes. The meta-analysis reveals that genes encoding major CVD risk proteins are highly expressed in the kidney, liver, and lungs. This indicates that vascular aging is inextricably linked to systemic organ failure and peripheral SASP burdens.
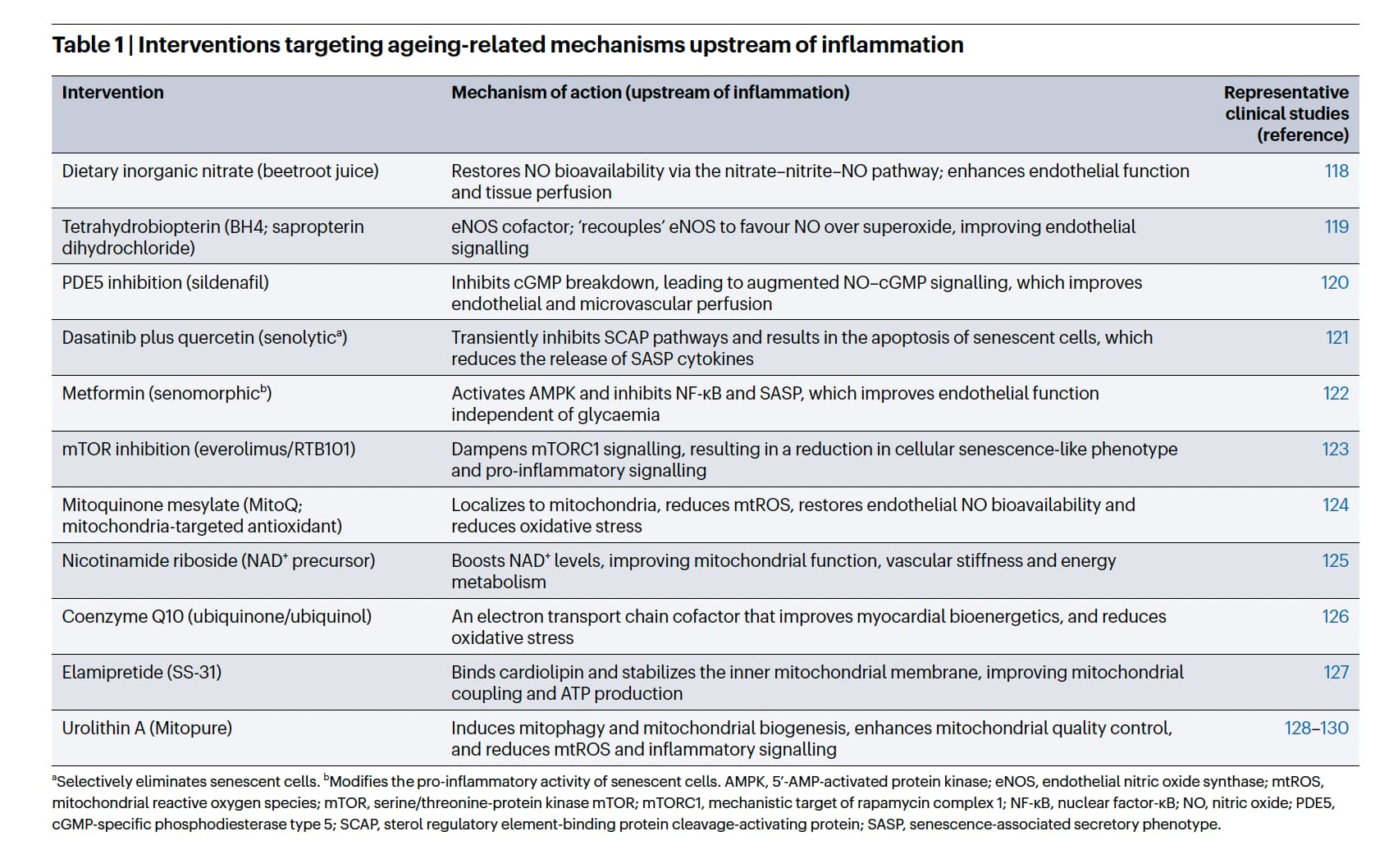
8. How do NO-boosting interventions (like PDE5 inhibitors) address the root cause of macromolecular damage rather than just temporarily masking the hemodynamic resistance? By dilating arterioles and improving microvascular tissue perfusion, PDE5 inhibitors restore the flow of electrons to oxygen in the mitochondria. This alleviates the reductive stress and ROS leakage that directly initiates the inflammatory cascade.
10. Does chronological age dictate the point of no return for these resilience mechanisms, or is the “entropic threshold” entirely determined by biological damage? It is entirely driven by biological entropy. While chronological age correlates with damage, the threshold at which resilience mechanisms fail and transition into maladaptive inflammation is dictated by the cumulative burden of stochastic cellular stressors, environmental insults, and un-repaired molecular damage.