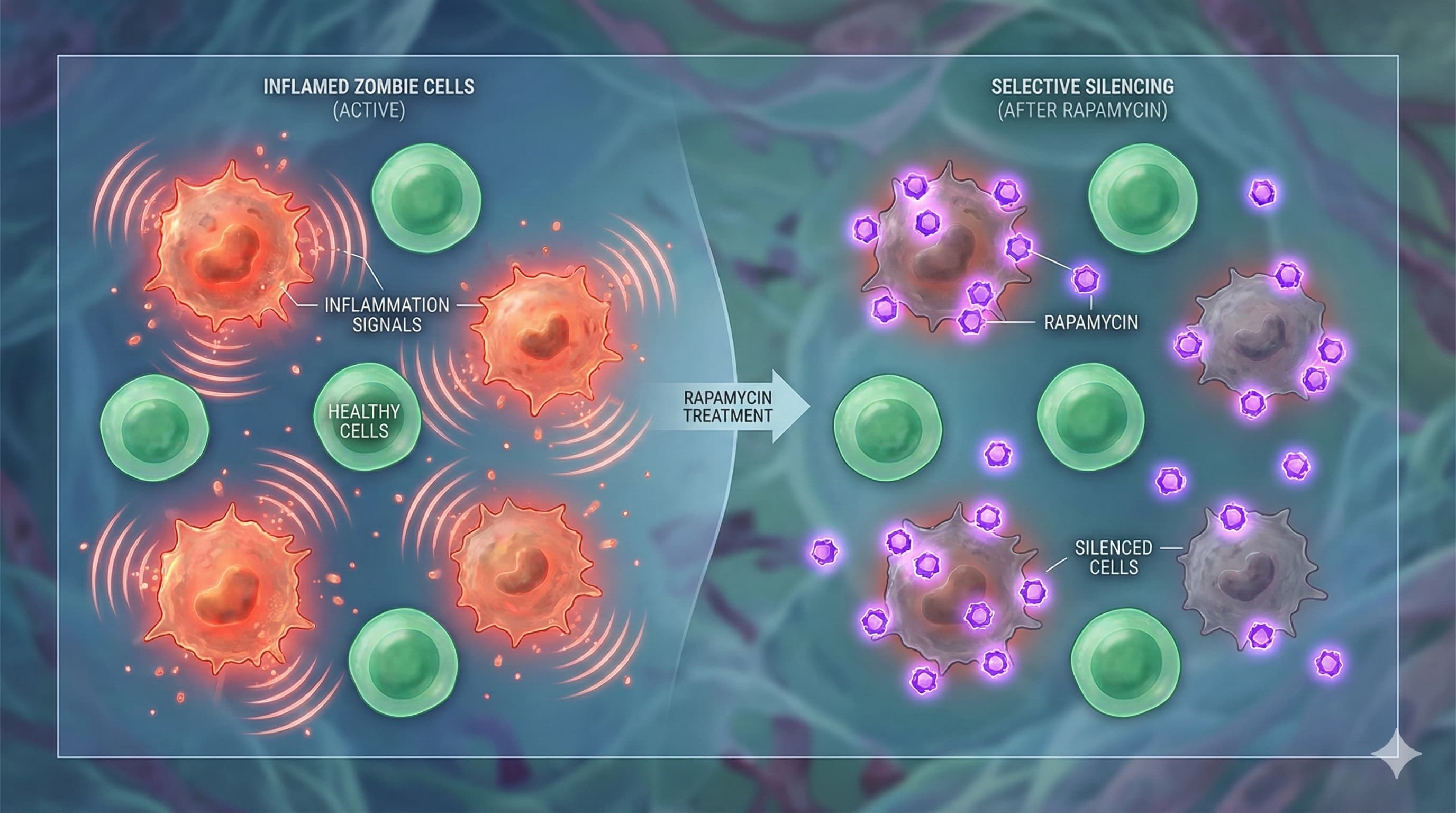
The binary view of cellular aging—that a cell is either healthy or “senescent”—is increasingly viewed as a dangerous oversimplification. New research from the University of Tasmania and Monash University, Australia, published in the top-tier journal Aging Cell, suggests that “zombie cells” exist on a pathogenic spectrum, and that the longevity drug rapamycin works by specifically targeting the most toxic sub-populations among them.
Using high-throughput single-cell fluorescence imaging on human fibroblasts, the researchers demonstrated significant heterogeneity in classical senescence biomarkers, including p21, SA-βgal, and IL-6. They discovered that nuclear morphology is a robust predictor of toxicity; cells with enlarged nuclei were highly correlated with elevated Interleukin-6 (IL-6) expression, a primary driver of the Senescence-Associated Secretory Phenotype (SASP). This confirms that not all senescent cells contribute equally to “inflammaging.”
Mechanistically, the study reveals that rapamycin does not act as a blanket senolytic (killing all senescent cells). Instead, it functions as a precision senomorphic. The drug selectively dampened the high-intensity biomarkers in the specific sub-populations characterized by enlarged nuclei and hyper-active SASP. This implies that rapamycin’s longevity benefits may stem from modulating the mTOR pathway to suppress the protein synthesis required for the toxic secretory phase of specific “super-senescent” cells, rather than simply halting the cell cycle or inducing autophagy universally. By silencing the “loudest” cells responsible for systemic inflammation, rapamycin effectively lowers the cumulative inflammatory burden without requiring total senescent cell clearance.
Actionable Insights for Longevity Biohackers
Refining the Rapamycin Protocol: The data supports the “senomorphic” use case for rapamycin. Rather than high-dose “shock” therapy intended to kill cells, consistent, modulation-focused dosing (e.g., weekly pulsed dosing) may be more effective at keeping the high-SASP sub-populations suppressed, preventing the systemic inflammatory cascade.
Morphological vs. Chemical Biomarkers: Standard commercial senescence tests often rely on aggregate SA-βgal. This study suggests this is insufficient. Biohackers conducting n=1 trials should prioritize circulating inflammatory markers (hs-CRP, IL-6, TNF-α) as proxies for the activity of senescent cells, rather than just their presence.
Vascular Health Proxy: Given that fibroblasts (connective tissue) were the model, monitoring pulse wave velocity (arterial stiffness) or endothelial function could serve as a functional readout for whether the suppression of fibrotic/senescent signaling is translating to tissue flexibility.
Cost-Effectiveness
ROI: High. Rapamycin (sirolimus) is a generic pharmaceutical. Given that this study highlights its ability to neutralize the most damaging subset of senescent cells without the need for expensive, novel senolytics, the marginal benefit per dollar is exceptional compared to proprietary peptides or experimental gene therapies.
Critical Limitations
In Vitro Constraints: The study utilized cultured human fibroblasts induced via chemotherapy (mitomycin C) and oxidative stress. This ignores the complex immune surveillance (NK cells, macrophages) present in a live organism that usually clears these cells.
Senomorphic vs. Senolytic Ambiguity: While the study shows biomarker suppression, it does not confirm if these “tamed” cells eventually undergo apoptosis or if they rebound immediately upon cessation of rapamycin, necessitating lifelong adherence.
Tissue Specificity: Fibroblast behavior may not translate to post-mitotic tissues like neurons or cardiomyocytes. The nuclear-enlargement correlation might be specific to structural cells.
“Inflammation and mitochondrial health are tightly intertwined. Protecting mitochondria (via lifestyle, metabolic management, and, in research settings, specific drugs/supplements) is one of the reasons so much attention is being paid to anti-inflammatory strategies in aging and chronic disease.”
@John_Hemming would you agree with this statement from Gemini on this issue?
There is robust, high-confidence evidence from multiple other studies confirming that Rapamycin directly induces mitophagy. This mechanism is considered one of the primary drivers of its longevity and neuroprotective effects.
1. Mechanism of Action
mTORC1 Inhibition: Rapamycin inhibits mTORC1, which is a negative regulator of autophagy. Under normal nutrient-rich conditions, mTORC1 phosphorylates and inhibits ULK1 (a mitophagy initiator). By blocking mTORC1, Rapamycin releases the “brake” on ULK1, allowing it to initiate the encapsulation of defective mitochondria into autophagosomes.
PINK1/Parkin Pathway: Research indicates Rapamycin enhances the recruitment of the protein Parkin to damaged mitochondria, a critical step in tagging them for destruction.
2. Key Supporting Studies
Mitochondrial DNA Quality (Mouse Muscle): A landmark study (PMC5911406) showed that long-term Rapamycin treatment in aging mice significantly reduced the frequency of mitochondrial DNA (mtDNA) deletion mutations.
Mechanism: The drug did not “repair” DNA; rather, it stimulated mitophagy to selectively clear the mitochondria carrying high loads of mutated DNA, effectively “purifying” the cellular mitochondrial pool.
Neuroprotection (Alzheimer’s Models): In mouse models of Alzheimer’s (e.g., APP/PS1 mice), Rapamycin was shown to restore cognitive function specifically by enhancing mitophagy (Source: The Journals of Gerontology , 2021). The study used fluorescence markers to visually confirm the fusion of mitochondria with lysosomes (mitolysosomes) following Rapamycin treatment.
Mitochondrial Myopathy: In cases of severe mitochondrial disease (Cox15 mutants), Rapamycin rescued muscle function not by fixing the genetic defect, but by inducing autophagy/mitophagy to clear the organelles that were producing the most toxic byproducts (ROS).
Summary for the Biohacker
While the Seshadri paper tells you “who” Rapamycin targets (the large-nucleus, high-inflammation cells), the broader literature tells you “how” it likely works: by triggering mitophagy to clear the metabolic junk (damaged mitochondria) that is driving that inflammation.
Takeaway: The “senomorphic” effect observed in the Seshadri paper—where cells are “tamed” rather than killed—is highly consistent with a burst of mitophagy that cleans up the cell’s internal environment, lowering oxidative stress (ROS) and shutting off the IL-6 alarm signal.
I don’t know how Gemini works. Is it giving us the correct answer which seems to make sense to me, or is it finding an easy explanation? Now, if only there was some human data.
That far too strong of a statement. mTOR is found in various parts of cells, including the nucleus and cytoplasm. Therefore rapamycin is bound to effect various parts of the cell, mitochondria being just one of those. To claim that its effects on mitochondria are much more important than its effects on other parts of cells requires strong reasoning or proof (such as a lifespan study showing that inhibiting mitophagy blocks the effects of rapamycin on lifespan).
I understand. I just think that phrasing like “In the end it is the mitochondria that matter.” is stronger than what the current evidence establishes, unless you mean it as a personal way of thinking about the problem rather than an actual conclusion.
I consider in the round it to be a conclusion, but I don’t think it helps to debate the nuances of wording. Over time there will be experimentation and additional interventions produced. This will lead to greater certainty about what affects what. With all my reading and experimentation I have personally concluded that there are two biochemical statuses that really matter that is the state of the mitochondria (via mtDNA) and the presence of senescent cells (because they produce SASP which is pro aging).
It is worth trying to resolve disagreements about factual issues which have a bit more precision, but the agreed facts are insufficient to resolve this particular question of nuance.
I’m of course not against you having a personal theory of aging and of what is going on in the case of rapamycin use. I’m objecting to you presenting your theory as conclusion.
It’s almost impossible to establish causal effects with n=1 self experiments especially when you cannot even measure mitochondrial health directly with the tools you are using. Even if you notice some changes, you cannot reasonably conclude they were primarily caused by effects on mitochondria rather than any number of other possibilities.
When you say “in the end it’s the mitochondria that matter,” that’s a strong causal claim. Given that rapamycin modulates multiple systems, attributing its benefits primarily to mitochondria would require evidence that mitochondrial effects are necessary and/or dominant relative to the other pathways (e.g., interventions or models where mitochondrial mechanisms are selectively perturbed and rapamycin’s benefits diminish accordingly). We don’t have that.
I’m not saying your theory is impossible, just that the current evidence supports it as a hypothesis not a conclusion.
Btw even when I see a clear n=1 effect, I treat mechanism as hypothesis. As an example. High dose chromium markedly improved my blood glucose decades ago. There are reasonable mechanisms by which it could have done so (as seen in animal an human studies) but I cannot conclude what truly happened in my case since I simply don’t have tools to measure what’s going on in my body intracellularly. So I am comfortable stating that chromium helped but not that I know how, I could just hypothetize how. The same logic applies to your claim about rapamycin actiing through mitochondria or that mitochondria are that matters. I keep pushing on this here because in science, acting overly certain about mechanisms of action is a fast way to lose credibility.
You are not actually using any substantive arguments here. You are merely using your opinion against mine. I will, however, leave it after this. If you simply contradict my opinions without evidence I will just ignore it so that we don’t both waste other people’s time reading this.
Whether or not I’ll live few years longer, I think its anti-inflammatory effects are as good as those of the pain-relieving peptides and significantly better than acetaminophen and ibuprofen. This alone makes it a great addition to my personal pharmacy.
This isn’t opinion vs opinion. You made a strong claim of causal primacy (“mitochondria are what matter in the end”), which carries a burden of evidence. I’m pointing out that we don’t currently have evidence establishing mitochondrial mechanisms as being the necessary or dominant part of rapamycin’s benefits, and that n=1 self-experiments can’t resolve that.
We must not forget that one of the most important standards in any serious scientific discussion is to remain humble about ones knowledge and recognizing what one doesn’t know or cannot know with the current tools and evidence. That’s what I’m trying to say here and I think most of us here can agree on that.