I really like that model, Antoine. It has a lot of explanatory power and fits the data pretty well. If I were to go by intuition, I think it’s likely to be correct - but then again, I’ve had this kind of model turning in my head for a long time. It fits a lot, including the cholesterol/statin PD contradictions (which btw. I’m still working on, it’s a very deep rabbit hole, unfortunately).
2 Likes
I’m a cuddle monster!!
But yes, I do find some benefit to my demeanor, I’m not quite as mean LoL!
One of the things that Bryan J talks about is Nocturnal penile tumescence (NPT). This is an indicator of “age”. Over the last 5 years I’ve “noticed” it doesn’t seem to happen as much. But it has risen, to a more noticeable level in the past couple months. ![]()
3 Likes
Produced by ChatGPT 4.5 after feeding it the three papers ![]() I like it as well.
I like it as well.
1 Like
McGill + Northwestern + Cornell
Background: All the processes leading to neurodegeneration cannot be addressed with just one medication. Combinations of drugs affecting various disease mechanisms concurrently could demonstrate improved effect in slowing the course of Parkinson’s disease (PD).
Objective: This was a drug-repurposing experiment designed to assess several combinations of nine drugs for possible added or synergistic efficacy using in vitro models of PD.
Methods: We evaluated 44 combinations of the nine medications (sodium phenylbutyrate, terazosin, exenatide, ambroxol, deferiprone, coenzyme-Q10, creatine, dasatinib and tauroursodeoxycholic acid) selected for their previously demonstrated evidence of their impact on different targets, showing neuroprotective properties in preclinical models of PD. We utilized wild-type induced pluripotent stem-cell-derived human dopaminergic neurons treated with 1-methyl-4-phenylpyridinium for initial screening. We retested some combinations using an idiopathic PD patient-derived induced pluripotent stem cell line and alpha-synuclein triplication line. We assessed anti-neuroinflammatory effects using human microglia cells. As metrics, we evaluated neurite length, number of branch points per mm2, the number of live neurons, neurofilament heavy chain and pro-inflammatory cytokines.
Results: We have identified four combinations of two to three drugs that showed an additive protective effect in some endpoints. Only the combination of sodium phenylbutyrate, exenatide and tauroursodeoxycholic acid showed improvement in four endpoints studied.
Conclusions: We demonstrated that some of the medications, used in combination, can exert an additive neuroprotective effect in preclinical models of PD that is superior to that of each of the compounds individually. This project can lead to the development of the first treatment for PD that can slow or prevent its progression.
1 Like
Its all a question as to at what point you catch the process that causes PD.
Regarding this great paper: what happens first, DNA damage or alpha-syn aggregation?
Melatonin is interesting as it might protect from DNA damage and/or improve DNA damage repair capacity: Melatonin megadoses? - #295 by adssx
And there’s some evidence in PD: Parkinson's disease - #679 by adssx
Also:
- α-Synuclein reduces acetylserotonin O-methyltransferase mediated melatonin biosynthesis by microtubule-associated protein 1 light chain 3 beta-related degradation pathway 2024: “Our findings showed that α-SYN reduced the level and activity of melatonin synthesis enzyme acetylserotonin O-methyltransferase (ASMT) in the pineal gland and in the cell cultures. In addition, we found that microtubule-associated protein 1 light chain 3 beta (LC3B) as an important autophagy adapter is involved in the degradation of ASMT. […] One possible reason of this mitigative effect is that injection of melatonin reduces the toxic α-SYN in the important RBD brain tissues. We detected the melatonin level and the α-SYN level in the circadian clock regulation center (hypothalamus), the results showed that melatonin is decreased in the hypothalamus of TG mice, which can be increased after injection of melatonin. While the protein level of α-SYN is nearly unchanged before or after injection of melatonin. These results tend to that the changed RBD-like behaviors in TG mice after melatonin injection are a result from a rescuing effect of melatonin. However, these results in other important RBD brain tissues remain to be further explored. […] Although melatonin affects the α-SYN induced RBD-like behaviors, the photo-instability of melatonin determines its limitations in the induction of these behaviors, indicating that there may be other important molecules in the process. For instance, the hypocretin neurons control motor during wakefulness and sleep in humans, their deficiency induces a motor control defect during REM sleep”
- Melatonin MT1 receptors regulate the Sirt1/Nrf2/Ho-1/Gpx4 pathway to prevent α-synuclein-induced ferroptosis in Parkinson’s disease 2024: “Our findings reveal a novel mechanism by which MT1 activation prevents α-syn-induced ferroptosis in PD, highlighting the neuroprotective role of MT1 in PD.”
- Microglial Melatonin Receptor 1 Degrades Pathological Alpha-Synuclein Through Activating LC3-Associated Phagocytosis In Vitro 2024: “Taken together, the results suggest the neuroprotective function of microglial cells in clearing α-Syn through MT1-mediated LAP, highlighting the potential key role of MT1 in pathogenic mechanisms associated with α-Syn.”
- Modulation of autophagy by melatonin and its receptors: implications in brain disorders 2024
- Effect of melatonin on α-synuclein self-assembly and cytotoxicity 2012: “Initial studies revealed that Mel blocked αS fibril formation as well as destabilizing preformed αS fibrils. Subsequent evaluation of the assembly-stage specificity of the effect showed that Mel was able to inhibit protofibril formation, oligomerization, and secondary structure transitions. Importantly, Mel decreased αS-induced cytotoxicity. These data suggest a mechanism of action for Mel, inhibition of assembly of toxic polymers and protection of neurons from their effect.”
2 Likes
Doctor Behind Award-Winning Parkinson’s Research Among Scientists Purged From NIH
“Multiple sources at the NIH, granted anonymity because they were not authorized to talk to the media, confirmed Tuesday afternoon that at least 10 principal investigators who were leading and directing medical research at the agency had been fired. Among them is Dr. Richard Youle, a leading researcher in the field of neurodegenerative disorders previously awarded the Breakthrough Prize in Life Sciences for his groundbreaking research identifying mechanisms behind Parkinson’s disease.”
Given how long it takes to establish research infrastructure, this development is not good. Even if these scientists get work at other institutions immediately, it takes time to set up new studies, set goals, get the necessary collaborators and so on.
And getting fired means that whatever research program they were in the middle of, is now disrupted or collapsed.
Neurodegenerative disease studies take a long time. It is unfortunate that a lot will now be set back. Rates of PD are increasing and we need to tackle this as soon as possible.
I am very sad and disappointed with these developments. I always thought of medical research as a steadily growing endeavor. I didn’t expect that as the 21st century is progressing, we’ll actually retrench our efforts, just as the needs of an aging population grow. Pretty discouraging. I don’t understand it - we’re all in this together, including those who have made these decisions, medical science affects their and their families lives too. Who thinks, “whelp, I and my family don’t need medical science, we’re good.” SMH.
3 Likes
Sometimes you have to wonder what some people are thinking.
Personally, I think those people aren’t thinking at all.
This is a horrible loss to humanity.
1 Like
Targeting mitochondria-regulated ferroptosis: A new frontier in Parkinson’s Disease therapy 2025
Mitochondrial dysfunction is a key driver of ferroptosis in PD.
Altered mitochondrial function affects ETC, TCA, and mLIP to promote ferroptosis.
Ameliorating mitochondrial damage-induced ferroptosis holds a great promise for the therapeutic approach for PD
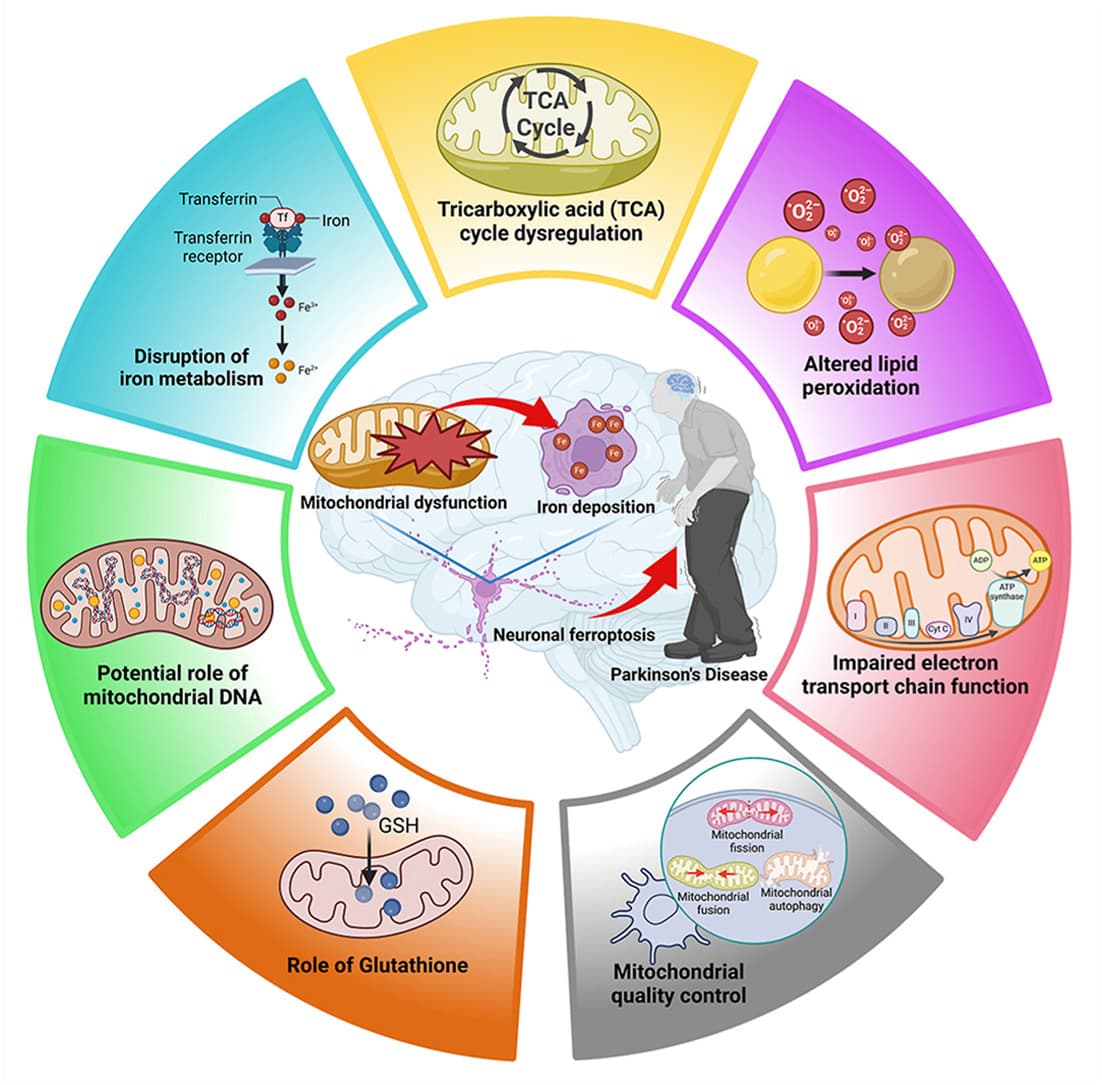
1 Like
I intend writing up a post about melatonin and the levels required to protect neurons some time today.
I have started writing this up, but I think it will take a few days to bring together all the separate research.
4 Likes
Not sure if this is relevant to Parkinson’s specifically:
Intermittent fasting and neurocognitive disorders: What the evidence shows
Conclusion: Current findings highlight the therapeutic potential of IF for individuals with existing cognitive impairment. While preclinical studies provide robust evidence of neuroprotective mechanisms, human studies remain sparse and require standardization. Further clinical research is necessary to confirm long-term safety and efficacy and to refine IF protocols for broader clinical application.
2 Likes
Previous studies have reported no significant differences in circadian phase, as measured by DLMO, between people with PD and healthy controls. However, amplitude of markers of circadian rhythms are blunted. Videnovic and colleagues highlighted significantly reduced melatonin amplitude and melatonin area under the curve throughout the day in people with PD, compared to controls, despite no differences in DLMO timing. Similarly, Bolitho and colleagues observed that circadian phase remained unchanged between PD and control groups.
Comparable findings were evident in relation to the circadian rhythm of core body temperature. While no phase variations have been presented, core body temperature appears to be disrupted in people with PD. Zhong and colleagues highlighted that PD patients have a reduced average core body temperature in conjunction with a blunted nadir, when compared to healthy controls. The reduction in the amplitude of core body temperature and melatonin rhythms, rather than a phase shift (such as delayed or advanced timing), indicates that PD-related circadian disruptions may manifest as weaker or dampened circadian signals rather than as complete phase shifts.
Like dementia and AD, it appears that people with PD experience disrupted rest-activity rhythms. However, the disruption has been specifically associated with disease severity. In early and moderate stages of PD, rest-activity rhythms are preserved, despite reduced overall activity levels compared to healthy controls. Whereas in advanced stages of PD, circadian variation diminishes and activity patterns become more fragmented. Furthermore, alteration in rest-activity rhythms appear to be influenced by common pharmacological treatments. These findings suggest that both disease progression and medication effects contribute to the disruption of rest-activity rhythms in PD.
Due to the growing evidence of an association between circadian rhythm disturbances and cognitive and functional decline, it has been suggested that circadian dysfunction could contribute to early pathogenesis and serve as a surrogate marker of neurodegenerative diseases. Interventions aimed at stabilising these rhythms could potentially mitigate the risk of AD, dementia and PD, and enhance cognitive health in older adults.
Caffeine increases the oral bioavailability of melatonin: Effects of caffeine intake on the pharmacokinetics of melatonin, a probe drug for CYP1A2 activity - PMC
Smoking seems to decrease melatonin levels.
And yet both caffeine and smoking are protective against PD. So this would argue against low melatonin being causal @John_Hemming ? (unless serum levels don’t matter and only the CSF matters and caffeine and smoking don’t impact it?)
A bit more complex, per ChatGPT:
However, research findings on this topic have been mixed. Some studies have reported decreased blood melatonin levels in active smokers, suggesting that smoking may lead to lower circulating melatonin. Conversely, other studies have found higher daytime circulating melatonin levels in smokers compared to nonsmokers, indicating a complex relationship that may depend on various factors such as timing of measurement and individual differences.
The serum levels don’t really matter for PD. My hypothesis is that it is the higher CSF levels that are needed. These are really very high compared to serum levels.
Research on SLU-PP-332 and related ERR modulators continues to expand into new therapeutic areas:
- Neurodegeneration: Preliminary studies suggest potential applications in Alzheimer’s and Parkinson’s diseases through improved mitochondrial function
1 Like
World-first trial indicates immunosuppression may help treat Parkinson’s
Participants on the treatment reported an overall improvement in movement-related symptoms. This meant they found it easier to perform tasks such as moving around, writing, washing and dressing. These improvements were greater in female participants compared to males.
Additionally, patients with faster-progressing Parkinson’s disease showed signs of improved memory and thinking skills.
Importantly, the trial did not reveal significant safety concerns around using immunosuppression treatments in Parkinson’s disease This will enable larger research studies of immune therapies for PD in the future.
During the trial, the effect of the drug on the immune system was measured in both the blood and brains of participants. It showed that, although Azathioprine cannot cross the blood-brain barrier, it acts indirectly to slow progression of inflammation in the brain, and this could explain its effects in Parkinson’s disease.
Very interesting @DrFraser @John_Hemming: positive effects of immunosuppression despite no BBB crossing!
This might explain the gender difference: Parkinson's disease - #629 by adssx
Details of the trial: ISRCTN
- 67 participants
- The treatment will be started at a low initial dose of 1 mg/kg.
- At four weeks, the dose of the trial drug will be increased, if appropriate according to the clinical profile (blood results and adverse events). Azathioprine will be increased to 2 mg/kg and the placebo dose will be doubled.
- “Their final visit will occur 6 months after the end of treatment. The MDS-UPDRS III will be performed in the OFF state, and videoed. Immediately following this the participant can take their PD medication. The remainder of the MDS-UPDRS will then be performed. Their medical and medication history will be reviewed. They will complete the Addenbrookes Cognitive Examination-III. They will go through the same questionnaires related to depression, quality of life, activities of daily living and non-motor symptoms. Finally, they will have a blood sample taken.”
So they saw improvements after a 6-month washout period?! That’s impressive!
4 Likes
Alpha-synuclein regulates nucleolar DNA double-strand break repair in melanoma 2025
Although an increased risk of the skin cancer melanoma in people with Parkinson’s disease (PD) has been shown in multiple studies, the mechanisms involved are poorly understood, but increased expression of the PD-associated protein alpha-synuclein (αSyn) in melanoma cells may be important. Our previous work suggests that αSyn can facilitate DNA double-strand break (DSB) repair, promoting genomic stability. We now show that αSyn is preferentially enriched within the nucleolus in melanoma, where it colocalizes with DNA damage markers and DSBs. Inducing DSBs specifically within nucleolar ribosomal DNA (rDNA) increases αSyn levels near sites of damage. αSyn knockout increases DNA damage within the nucleolus at baseline, after specific rDNA DSB induction, and prolongs the rate of recovery from this induced damage. αSyn is important downstream of ataxia-telangiectasia–mutated signaling to facilitate MDC1-mediated 53BP1 recruitment to DSBs, reducing micronuclei formation and promoting cellular proliferation, migration, and invasion.
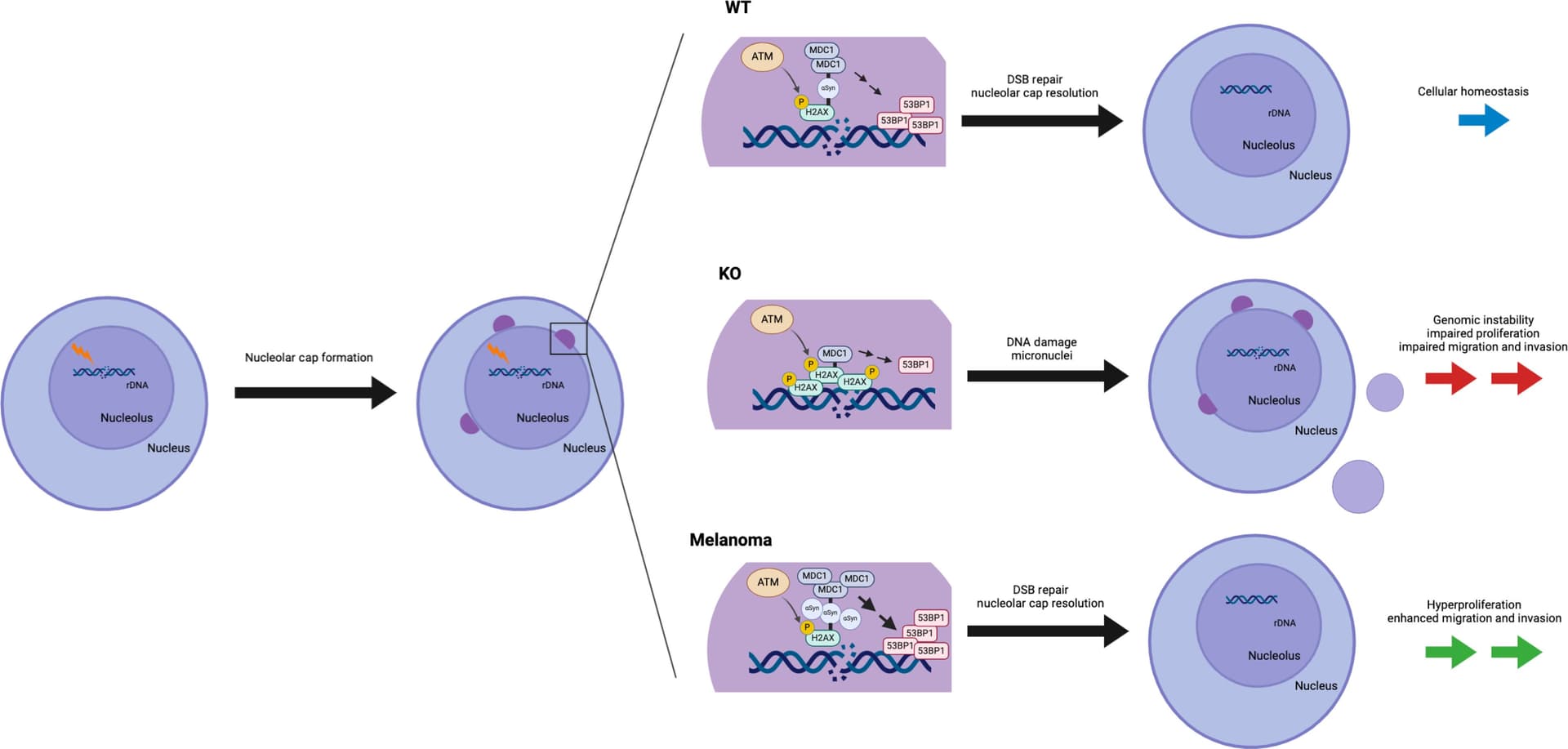
Our findings that αSyn modulates nucleolar DSB repair and that its loss of function negatively affects cellular growth suggest that up-regulation of αSyn levels in melanoma may also be part of a similar mechanism to improve DSB repair, allowing these cells to evade the programmed cell death and senescence pathways that would normally be triggered by high DSB levels. In contrast to what is seen in neurons, the high overexpression of αSyn by melanoma cells does not lead to frank aggregation or the formation of detectible Lewy pathology within these cancer cells. The reasons for this are unclear but could involve melanoma-specific factors that act to limit this protein’s aggregation and allow it to remain soluble, even when it is at high concentration within the cell. Alternatively, if melanoma cells were to form Lewy pathology and then die quickly, then they would also be hard to detect in a melanoma sample at any one given point in time.
It is well established that patients with PD and their first-degree relatives are at increased risk of melanoma and, symmetrically, that patients with melanoma are at increased risk of PD (1, 4–19, 22–24). Our previous work suggests that genetic or environmental factors that cause increased αSyn expression within certain individuals would predispose their postmitotic neurons to accumulate cytoplasmic Lewy pathology, and this, counterintuitively, triggers a loss of soluble, functional αSyn from the nucleus. This could lead to deficient DSB repair that contributes to programmed cell death (35, 36). Our current data suggest that these same individuals could also be predisposed to develop melanoma via a gain-of-function mechanism where increased αSyn levels improve DSB repair capacity within the nucleolus, which limits the senescence and programmed cell death pathways that are triggered by excessive DSBs associated with oncogenesis. This provides a framework for understanding the link between PD and melanoma and offers potential therapeutic targets in melanoma that are focused on reducing αSyn-mediated nucleolar DSB repair.
Reminder, it’s a dangerous drug:
I tend to think that the genome does quite complicated things and that when it goes wrong it will go wrong in complicated ways. I take a sort of computer debugging approach which is to fix the things we know are wrong and then see what problems remain.
Research suggests a potential link between varicella zoster virus (VZV) and Parkinson’s disease (PD), but the causal relationship between anti-VZV IgG levels and PD is not well understood. Using two-sample Mendelian Randomization (MR), we assessed the causal impact of anti-VZV IgG levels on PD risk and progression. Our study found a significant association between higher anti-VZV IgG levels and an increased risk of PD. For PD progression, higher anti-VZV IgG levels were linked to a greater risk of constipation, insomnia, and Restless Legs. These findings remained consistent after sensitivity analyses. In conclusion, our study suggests that elevated anti-VZV IgG levels may contribute to an increased risk and progression of PD, supporting a potential causal link that warrants further mechanistic investigation.
Our MR analysis indicated a significant association between increased anti-VZV-IgG levels and a higher risk of PD (IVW OR = 1.1388, 95% CI 1.0146–1.2781, P = 0.0273). This suggests that past exposure to varicella zoster virus may influence the risk of PD. Our findings align with meta-analytic evidence demonstrating a 20% increased risk of PD among individuals exposed to pathogens, including hepatitis C virus (HCV) and influenza A35. These pathogens share common mechanisms such as blood–brain barrier disruption, neuroinflammation, and direct neuronal damage. However, our study extends this literature by specifically implicating varicella-zoster virus (VZV) through elevated anti-VZV IgG levels as a potential risk modulator.
While our MR analysis supports a causal association between anti-VZV IgG levels and PD, it does not directly establish a biological mechanism. Future studies should explore whether VZV reactivation triggers α-synuclein aggregation, disrupts dopaminergic neurotransmission, or induces chronic neuroinflammation—pathways implicated in other viral infections. Experimental models (e.g., VZV-challenged neuronal cultures or animal studies) are needed to validate these hypotheses. If VZV-specific mechanisms are confirmed, antiviral prophylaxis (e.g., zoster vaccination) could be prioritized in high-risk populations, paralleling strategies proposed for HCV-related PD risk reduction.
Several limitations must be considered. First, while anti-VZV-IgG levels provide insight into past VZV exposure, they do not distinguish between the virus itself and the immune response it triggers. Therefore, it is unclear whether the VZV virus directly causes PD or if the immune response contributes to disease development. Second, the study lacked GWAS data for anti-VZV-IgM, which could have provided additional insights. Third, the statistical power to detect smaller effect size changes may be limited, and sample overlap between the GWAS datasets for anti-VZV-IgG and PD could introduce bias41. However, the high mean F-statistic of the SNP set suggests minimal impact from sample overlap42. Further studies in diverse populations are needed to confirm these findings and explore the causal relationship between anti-VZV-IgG levels and PD.