Well, pioglitazone failed against PD (I posted a study to that effect some time ago) - wrt ppar I have more faith in high dose telmisartan, but needs proving. I am waiting on ambroxol (and waiting, and waiting ![]() ), but while there are a lot of mechanistic reasons for hope, hasn’t it already failed once? DHA-rich omega-3: no thank you (at least as far as exogenous supplementation with DHA). LLT - statins - are a mixed bag, should work, but really doesn’t quite do it. I posted that paper above, but I really am not sure what is actionable here. I was hoping someone more versed in lipid physiology would chime in, I myself am not super versed in this. The only way I’ve looked more closely at lipids is in the context of calcium channels (lipid microdomains/rafts), and it’s clear that perturbed lipid metabolism results in features of NDDs including through modulating calcium channel signaling. But there is a lot that’s still not understood, and I myself know even less toward the lipid end of this - I think this paper is more exploratory than anything.
), but while there are a lot of mechanistic reasons for hope, hasn’t it already failed once? DHA-rich omega-3: no thank you (at least as far as exogenous supplementation with DHA). LLT - statins - are a mixed bag, should work, but really doesn’t quite do it. I posted that paper above, but I really am not sure what is actionable here. I was hoping someone more versed in lipid physiology would chime in, I myself am not super versed in this. The only way I’ve looked more closely at lipids is in the context of calcium channels (lipid microdomains/rafts), and it’s clear that perturbed lipid metabolism results in features of NDDs including through modulating calcium channel signaling. But there is a lot that’s still not understood, and I myself know even less toward the lipid end of this - I think this paper is more exploratory than anything.
Thanks @adssx - I should mention of your punchlist, I am already also working through the following:
- Telmisartan 80mg/daily (I was taking losartan previously for hypertension, this substitutes for that)
- Shingles and tetanus both completed ~2 years ago
- Looking into lithium orotate
Will assess K2 and UDCA - both new to me. GLP-1 for me is tirzepatide/semaglutide. As others have stated, I figure at the worst, it can’t do any further damage and reducing inflammation is a net gain if not direct impact.
There are a range of different K2s. You need MK7 and possibly MK9, but MK4 may not do the job. There are other K2s, but they are not available as supplements.
Great that you’re looking at those!
Semaglutide does not cross the BBB at all and recently failed in the massive Alzheimer’s trial. Tirzepatide might be better. Liraglutide, dulaglutide and lixisenatide might cross the BBB more. Ongoing trials for PD for some of them. In the exenatide phase 3 trial in PD those on exenatide were slightly worse off than placebo although it wasn’t statistically significant.
Q10 failed in various PD trials btw. I’m not convinced by it.
Actually re-checked and it’s compounded TZ/dula, so maybe a bit better at BBB.
And CoQ10 for PD is firmly in my “a bonus if it actually helps” bucket. It’s several hundred lead bullets in lieu of a single silver one for quite some time to come, I think.
This is a very quick and simple overview, but the concept seems pretty powerful. Alkahest (the blood plasma company started by Tony Wyss-Coray at Stanford, has over 100 million blood samples and is now looking for early blood markers of diseases (it sounds like Parkinsons was one of the first that they started on) - so perhaps a way for early identification and disease process tracking…
I see red light was mentioned here a few years ago, but I’ll also share it was briefly mentioned in today’s Huberman podcast. He was interviewing the neuroscientist Glen Jeffery who very briefly touched on it helping the symptoms of Parkinson’s

A summary of the above video:
A. Executive Summary
The conversation presents a comprehensive argument that modern indoor light environments—dominated by LED-heavy short-wavelength spectra—constitute a chronic mitochondrial stressor, while long-wavelength light (red, near-infrared, ~650–900 nm) can acutely and chronically improve mitochondrial performance, visual function, and systemic metabolism. Glen Jeffery’s research program, spanning insects, mice, primates, and humans, supports the view that light is a key environmental variable shaping mitochondrial aging, on par with diet, movement, and temperature.
Jeffery shows that short-wavelength light depresses mitochondrial membrane potential and respiration in retinal and brain cells. In mice, daily exposure to LED-shifted spectra induces fat gain, fatty liver, elevated ALT, organ shrinkage (heart, liver, kidney), impaired glucose regulation, and abnormal sperm morphology—all without dietary changes. Mechanistically, this aligns with reduced ATP output, lowered mitochondrial reserve, and possibly elevated ROS and cGAS-STING activation.
Conversely, long-wavelength light accesses deep tissues (passing through skin, skull, and clothing) and interacts with nano-structured water around ATP synthase, reducing viscosity, increasing rotor speed, and inducing upregulation of respiratory-chain proteins. This produces two effects: (1) immediate ATP improvement, and (2) longer-term mitochondrial “repair mode.”
Human studies show:
• A ~20% reduction in glucose spike when red light is applied to a small patch of skin before an oral glucose load.
• A ~20% improvement in color-vision thresholds, lasting ~5 days, after 3 minutes of ~670 nm exposure to closed eyelids.
• Long-wavelength light reduces age-related rod loss, supporting slower retinal aging.
• Morning exposure is consistently more effective than afternoon exposure, revealing strong circadian coupling.
Jeffery argues that sunlight—rich in long wavelengths—is epidemiologically linked to lower all-cause mortality, while excessive LED exposure may represent an overlooked aging accelerator.
B. Bullet Summary
- Short-wavelength/blue-enriched LEDs blunt mitochondrial membrane potential and respiration.
- LED exposure in mice induces metabolic syndrome phenotypes independent of diet.
- Long-wavelength light penetrates skin, skull, and clothing; scattering distributes energy through tissues.
- Nano-confined water, not cytochrome c oxidase, is the dominant absorber at long wavelengths.
- Long-wavelength light increases ATP synthase rotor speed and respiratory-chain protein expression.
- Mitochondria operate as a body-wide community, signaling across distant tissues.
- Red/NIR exposure reduces mitochondrial-triggered apoptosis.
- A small illuminated skin area alters systemic glucose handling (~20% spike reduction).
- Abdomen illumination reduces Parkinsonian degeneration in primate models.
- Long-wavelength light slows retinal photoreceptor loss in aging animals.
- In humans, 3 minutes of 670 nm improves color-vision thresholds by ~20% for 5 days.
- Eye exposure works through closed eyelids; dose threshold behaves like a binary “switch.”
- Morning exposure yields far stronger benefits than afternoon exposure.
- Sunlight includes broad long-wavelength content absent in most indoor environments.
- Epidemiology links higher sunlight exposure to lower all-cause mortality.
D. Claims & Evidence Table
| Claim | Evidence Presented | Assessment |
|---|---|---|
| LED/blue light impairs mitochondrial function | Real-time mitochondrial imaging in mice; LED vs full-spectrum comparisons | Moderate–strong (animal data strong; human data limited) |
| LEDs promote metabolic dysfunction | Mouse studies showing fat gain, fatty liver, ALT elevation under identical diet | Moderate (requires replication & mechanistic isolation) |
| Long-wavelength light improves systemic glucose handling | Human OGTT study: ~20% reduction in glucose peak after back illumination | Moderate (small sample but clear effect) |
| Long-wavelength light improves color vision in humans | Multiple human tests showing ~20% improvement lasting 5 days | Strong (replicated, consistent) |
| Red/NIR reduces neurodegeneration in Parkinsonian primates | Abdomen illumination reducing symptoms | Moderate (small N, model validity reasonable) |
| Long-wavelength light penetrates skull, skin, clothing | Radiometry/spectrometry measurements; imaging through hands & skulls | Strong |
| Long-wavelength light interacts with nano-water, not cytochrome c oxidase | Water absorption spectrum matches effective wavelengths | Speculative–plausible (needs biophysical confirmation) |
| Morning superiority for mitochondrial effects | Cross-species circadian comparisons | Moderate |
| Sunlight exposure lowers all-cause mortality | Epidemiology from Sweden & UK (e.g., Weller et al.) | Moderate (observational, confounded) |
E. Actionable Insights (8 items)
- Morning long-wavelength exposure (within 1–3 hours of waking) is optimal; 3–10 minutes is sufficient to test effects.
- Avoid reliance on blue-heavy LEDs in primary living spaces; incorporate halogen/incandescent or daylight-spectrum lamps.
- Use red/NIR for pre-meal glucose control testing: illuminate a small torso patch for 5–10 min before standardized carb intake; track CGM metrics.
- For retinal aging metrics, try 3 minutes of ~670 nm through closed eyelids once every ~5 days; assess color contrast thresholds.
- Increase natural sunlight exposure, emphasizing morning and avoiding sunburn; monitor sleep, mood, and glucose over weeks.
- For indoor environments, prioritize broad-spectrum, high-CRI lighting with IR content; avoid IR-blocking glass where possible.
- Combine morning outdoor light with low-intensity aerobic work (walks), testing additive mitochondrial effects.
- Avoid “indiscriminate blasting” with high-power devices; low-irradiance, well-characterized red/NIR sources are safer.
H. Technical Deep-Dive
Mechanistic Axes
- Mitochondrial membrane potential (Δψm): Blue/short-wavelength light reduces Δψm, lowering ATP production and increasing mitochondrial distress signaling.
- Nano-water viscosity: Long-wavelength absorption reduces viscosity around ATP synthase, increasing rotor torque and ATP output.
- Mitochondrial protein upregulation: Long-wavelength exposure increases expression of ETC complexes (I–IV, V).
- Apoptotic threshold modulation: Red/NIR reduces cytochrome c release probability, delaying apoptosis.
- Circadian modulation: Mitochondrial proteome and ATP output are highest in the morning, explaining time-of-day dependent responses.
- Systemic “mitochondrial community” signaling: Energy-demand shifts, cytokine release, microvesicle communication, and redox changes allow distal effects (e.g., abdomen illumination → brain, skin → systemic glucose).
- Aging pathways: By improving Δψm and lowering mitochondrial distress, red/NIR plausibly reduces cGAS-STING activation, dampens inflammaging, and may shift AMPK/mTOR toward repair, although not directly shown in humans.
I. Fact-Check of Important Claims
| Claim | Consensus View | Verdict |
|---|---|---|
| Blue/LED light damages mitochondria | Animal data support mitochondrial impairment at high blue doses; human data incomplete | Partially supported |
| Red/NIR boosts ATP via water | Water absorption at these wavelengths is true; ATP synthase viscosity mechanism is plausible but unproven in vivo | Speculative, not disproven |
| Red/NIR reduces Parkinson’s degeneration | Photobiomodulation shows promise but mechanisms uncertain; human evidence preliminary | Weak–moderate |
| Light passes through body & skull | Biophysics supports deep penetration of NIR; well-established | Strong |
| Sunlight increases longevity | Observational studies (Sweden, UK) suggest correlation; causation unproven | Moderate |
| Red/NIR acutely improves human glucose handling | Small controlled study → effect appears real; needs replication | Moderate |
| Daily LED exposure contributes to NAFLD/obesity | Animal studies robust; human evidence indirect | Moderate but tentative |
Not sure if anyone has posted this already but if not, thought @adssx would find it interesting:
@medaura, thanks, from this article: “Nearly every scientist interviewed for this story does a few simple things. They filter their water, they run an air purifier, they don’t microwave plastic”
@John_Hemming on melatonin and PD (Chinese preprint): Regulation of mitochondrial dynamics and function by melatonin type 1 receptor in parkinson’s disease 2025
interesting…my father was an avid golfer and developed Parkinsons
This is quite probably one of the easiest things we do 3 to 4 times a week.
tVNS with a simple setup works. $100 and have the most flexible tVNS system on the market.
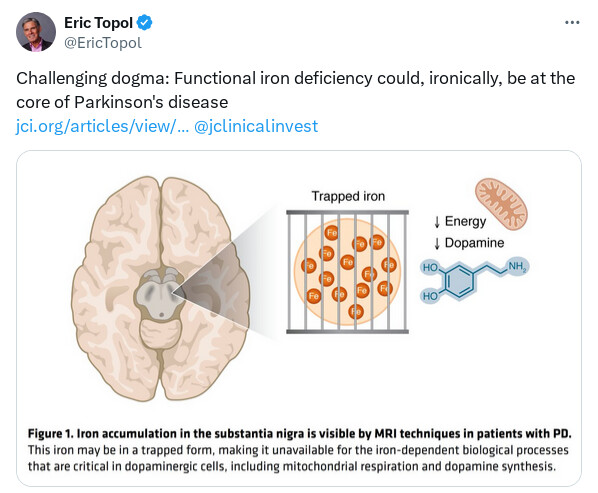
In conclusion, multiple lines of evidence call into question the iron overload hypothesis in PD pathology. If we consider instead that cells in humans with PD may be suffering from a functional iron deficiency (4), much more of the available data make sense. Epidemiological studies link systemic anemia and recent blood donations to higher PD risk (29). TH function and mitochondrial respiration both rely on iron; thus, functional iron deficiency will lead to decreases in dopamine tone and loss of mitochondrial respiration (which ultimately will drive cell death), both of which are hallmarks of PD. Iron removal via chelation only exacerbates these problems, particularly in drug-naive patients where excess l-DOPA is not on board to mask a decline (1, 2). Iron therapy, in RLS and in PD, benefits patients (9, 10). Removing iron in brains of patients with PD via chelation has been sufficiently tested clinically, with negative effects on patient outcomes. We should now consider the alternative hypothesis of functional iron deficiency and how we might tackle it therapeutically.
Isn’t it ironic? Functional iron deficiency at the core of Parkinson’s disease pathobiology
They’ll have to explain why anemia is protective in PD: https://www.cell.com/heliyon/fulltext/S2405-8440(24)10889-4
And why an iron chelator recently succeeded a PD like condition (MSA).
Gemini explains these paradoxes by saying:
- Anemia causes low oxygen and activates HIF which prevents iron from getting “stuck”
- ATH434 worked in MSA by being a mild chelator that redistributes the “trapped” iron rather than just removing in
So fixing failed iron metabolism might be the solution. Gemini says Ambroxol might help as well.
I think the problem is that neurons that are part of a neurodegenerative diseases as in are in themselves starting to fail and failing will fail in a number of different ways. What needs to be identified is the initial cause of the failure rather than the different ways in which they fail.
The Mitochondrial Connection in Parkinson’s Disease
Abstract
Mitochondria are highly dynamic organelles with complex structural features that perform several essential cellular functions, including energy production by oxidative phosphorylation, regulation of calcium and lipid homeostasis, and control of programmed cell death. Given their critical role, alterations in mitochondrial biology can lead to neuronal dysfunction and death. Defects in mitochondrial respiration, especially in oxidative energy production, have long been thought to be implicated in the etiology and pathogenesis of Parkinson’s disease. However, given the multifaceted roles of mitochondria in health and diseases, the putative role of mitochondria in Parkinson’s disease likely extends well beyond defective respiration. As such, mitochondrial dysfunction represents a promising target for disease-modifying therapies in Parkinson’s disease and related conditions.
https://perspectivesinmedicine.cshlp.org/content/16/1/a041891.full
The Powerhouse Betrayed: Parkinson’s as a Mitochondrial Autoimmune Failure**
In a paradigm-shifting thesis from Cold Spring Harbor Perspectives in Medicine, researchers (Schon, Matheoud, & Przedborski) dismantle the outdated view of Parkinson’s Disease (PD) as merely a “dying dopamine neuron” disorder. Instead, they reframe PD—and by extension, significant aspects of brain aging—as a systemic collapse of mitochondrial quality control that triggers a lethal immune response.
The “Big Idea” here is not just that mitochondria stop making energy (the classical ATP deficit model). It is that dysfunctional mitochondria, when not cleared by mitophagy (the cellular recycling crew), become toxic “antigen factories.” The authors highlight the PINK1-Parkin pathway, a mechanism that tags damaged mitochondria for destruction. When this pathway fails—due to aging or genetic mutations—damaged mitochondria release ancient bacterial-like DNA and proteins into the cytosol. This triggers the cGAS-STING pathway and other innate immune alarms, fooling the body into thinking it is under bacterial attack. The result is chronic, sterile neuroinflammation that cooks neurons alive.
For the longevity enthusiast, this paper is a “smoking gun” linking mitochondrial health directly to immune aging (inflammaging). It suggests that the path to preserving brain function isn’t just “boosting energy” with simple fuels, but aggressively enhancing the clearance of metabolic waste. The implications extend beyond PD: if you can keep your mitochondrial garbage disposal (mitophagy) running, you may delay the onset of neurodegeneration indefinitely.
Context:
- Institution: Columbia University (Vagelos College of Physicians and Surgeons) & Université de Montréal.
- Country: USA / Canada.
- Journal: Cold Spring Harbor Perspectives in Medicine.
Impact Evaluation: The impact score of this journal is ~7.8 to 10.1 (JIF), evaluated against a typical high-end range of 0–60+, therefore this is a High impact journal. While not a generalist giant like Nature, it is a premier “Elite” venue for deep-dive mechanistic reviews that define the consensus in molecular medicine.
Part 2: The Biohacker Analysis
Study Design Specifications
- Type: Comprehensive Mechanistic Review (Synthesizing In Vivo, In Vitro, and Clinical data).
- Subjects: N/A (Review of murine models, Drosophila, and human post-mortem/genetic data).
- Lifespan Data: Discusses healthspan/disease onset rather than maximum lifespan extension. *Key Metric:*Prevention of dopaminergic neuron loss (neuroprotection).
Mechanistic Deep Dive
The authors dissect the failure of the PINK1-Parkin Axis as the central driver of PD. Here is the biohacker breakdown:
- The Sentinel (PINK1): In healthy mitochondria, PINK1 is imported and degraded. In damaged ones, import fails, and PINK1 accumulates on the outer membrane, signaling “DESTRUCTION REQUIRED.”
- The Executioner (Parkin): PINK1 recruits Parkin (an E3 ubiquitin ligase) to tag the organelle with ubiquitin chains, flagging it for the lysosome (autophagy).
- The Lethal Leak (cGAS-STING): When this clearance fails (aging/mutation), mitochondria leak mtDNA. The cell perceives this mtDNA as a viral invader, activating cGAS-STING, which pumps out inflammatory cytokines (IL-6, Type I Interferons).
- Autoimmunity: The paper discusses the controversial but compelling “Mitochondrial Antigen Presentation” (MitAP) theory—that cells might present mitochondrial proteins on their surface (MHC-I), inciting T-cells to attack neurons.
Organ-Specific Priority: Substantia Nigra (Brain) and Gut-Brain Axis (early mitochondrial failure often starts in the intestine).
Novelty
The paper moves the needle by cementing the link between Mitochondria and Immunity. It shifts the focus from “energy crisis” to “immune trigger.” It implies that anti-inflammatory drugs are band-aids, while mitophagy inducersare the actual cure. It also lends massive weight to the “intestine-first” hypothesis, where gut infection/stress triggers mitochondrial failure that propagates to the brain.
Critical Limitations
- Translational Gap: Most mechanistic proof comes from PINK1/Parkin knockout mice, which frustratingly do not develop overt PD symptoms unless stressed (e.g., by exhaustive exercise or infection). Human biology is far more sensitive to mitochondrial defects than mouse biology.
- Missing Data: There is a lack of rigorous human clinical trials proving that pharmacological induction of mitophagy (e.g., via Urolithin A) definitively slows PD progression, despite strong animal data.
- Effect Size Uncertainty: While genetic forms of PD are clearly linked to this pathway, “Sporadic PD” (90% of cases) has a more tenuous link to PINK1/Parkin, though mitochondrial dysfunction is universally present.