Mining transcriptomes from over 16,000 human and thousands of mouse samples, researchers find that aging corrupts the cell’s RNA-editing machinery in a consistent, damaging way they call “splicing degeneration” — and that this corruption is partially reversed by rapamycin and calorie restriction, positioning it as a candidate new hallmark of aging.
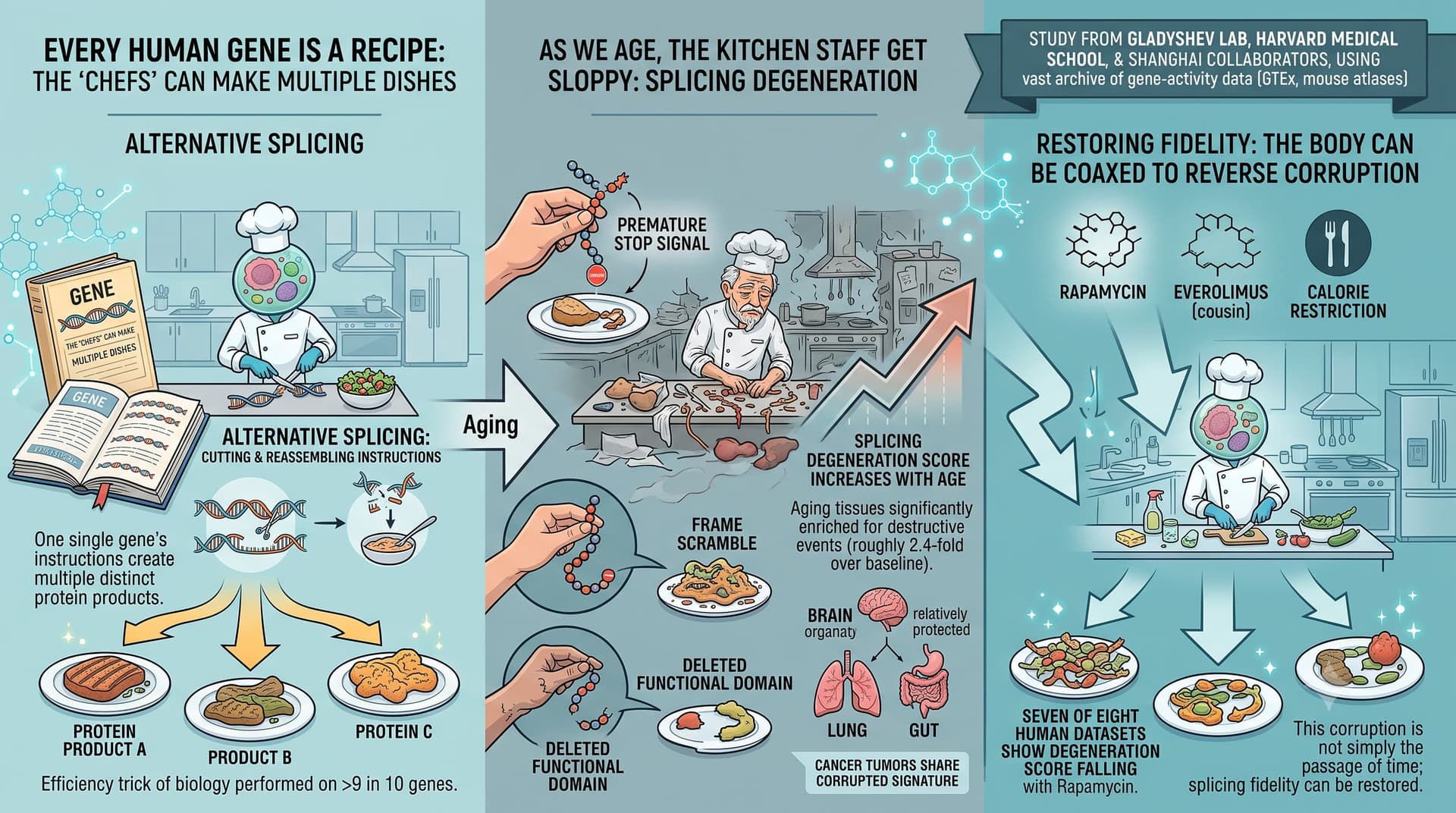
Every human gene is a recipe that can be cooked several ways. Through a process called alternative splicing, a single gene’s instructions are cut and reassembled into multiple distinct protein products — a feat performed on more than nine in ten of our genes. It is one of the great efficiency tricks of biology. This new study argues that, as we age, the kitchen staff get sloppy.
Researchers from Vadim Gladyshev’s lab at Harvard Medical School, working with collaborators in Shanghai, scoured a vast archive of gene-activity data: 16,627 human tissue samples from the GTEx project plus large mouse atlases. Rather than simply cataloguing which splicing patterns shift with age — which others have done — they asked a sharper question: do these shifts actually damage the resulting proteins?
Their answer is a fairly emphatic yes. The team built a classifier that flags splicing changes as “damaging” when they introduce a premature stop signal, scramble the protein’s reading frame, or delete a functional protein domain. Aging tissues, they found, are significantly enriched for exactly these destructive events (roughly 2.4-fold over baseline). They distilled this into a single “splicing degeneration” score that climbs steadily with age across most tissues. Intriguingly, the brain appears relatively protected, while the lung and gut are hit hard. Tumours, notably, show the same corrupted signature — hinting at shared machinery between aging and cancer.
The headline-grabbing part: the corruption is not simply the passage of time made visible. When the team examined cells and mice treated with rapamycin — the gold-standard lifespan-extending drug — seven of eight human datasets showed the degeneration score falling. Everolimus (a rapamycin cousin) and calorie restriction pointed the same direction. This suggests splicing fidelity is something the body can, in principle, be coaxed to restore.
Mechanistically, the authors finger specific “splicing factor” proteins — the regulators that decide how RNA gets cut — as culprits. Silencing many of these factors made cells look transcriptomically older on the lab’s own aging clocks, with the splicing-degeneration regulators producing the largest effect.
The big idea is to promote alternative splicing from a footnote in aging biology to a potential hallmark and therapeutic target in its own right. If splicing fidelity can be measured as a biomarker and nudged back toward youthfulness pharmacologically, it joins a growing list of aging processes that are, at least on paper, addressable. The caveat — and it is a large one — is that this entire edifice rests on correlation and computational inference, not on a single experiment showing that fixing splicing makes an animal live longer.
Actionable Insights
This is a biomarker-and-mechanism paper, not a trial. The “intervention” data are reanalyses of rapamycin, everolimus, calorie restriction, and metformin datasets that already informed longevity practice — the paper adds a new readout (splicing score), not a new intervention.
The take-home with the most evidential weight is that rapamycin’s benefit signal extends to a previously unmeasured layer of cellular fidelity — reinforcing its standing in a longevity stack. Effect-size reality check: the splicing-degeneration changes are statistically detectable but tiny in absolute terms. Treated-vs-control score shifts in the figures live in the third decimal place (e.g. ~0.235 → ~0.230), i.e. low-single-digit percent relative reductions. The strongest quantitative signal in the whole paper is the baseline enrichment of damaging splicing in old tissue (odds ratio 2.4), not the magnitude of any intervention’s reversal.
Practical translation for the stack-builder: nothing changes your protocol today. Rapamycin remains as a key supported lever; metformin and S6K1 deletion gave non-significant trends (p = 0.12 and 0.35). Do not treat “splicing degeneration” as a measurable personal biomarker — there is no assay you can order.
Source:
- Open Access Paper: Mammalian aging involves genome-wide splicing degeneration leading to functional decline, Posted June 29, 2026.
- Institutions: Division of Genetics, Brigham and Women’s Hospital / Harvard Medical School (Boston, USA); Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences (Shanghai, China)
- Country: USA / China (collaboration)
- Journal: bioRxiv — preprint server. Posted June 29, 2026. Not peer reviewed