Provoking title but interesting article:
Patients with Alzheimer’s disease (AD) have a decreased incidence of cancer, with a cross-sectional analysis of a nationwide sample of adults finding 21-fold higher odds of cancer diagnosis in non-AD individuals compared with those with AD. In this study, we demonstrated that mitochondrial localization of AD-associated amyloid-β precursor protein (APP) and its cleavage product amyloid-β 40, but not mutant APP that lacks a mitochondrial localization signal, inhibits lipid stress–mediated hyperactive mitophagy in aging T cells, improving their antitumor functions. Growth of melanoma xenograft or carcinogen-induced oral cancer models was highly reduced in AD mice. Additionally, adoptive cell transfer–based immunotherapy using aging T cells isolated from AD mice suppressed tumor growth. The metabolic signature of stress-dependent mitophagy in T cells showed fumarate depletion, which was linked to decreased succination of Parkin and enhanced mitochondrial damage. Mechanistically, APP interaction with the TOMM complex at the outer mitochondrial membrane attenuated trafficking of ceramide synthase CerS6 to mitochondria in aging AD T cells, preventing ceramide-dependent mitophagy. Thus, APP restored mitochondrial fumarate metabolism and Parkin succination, improving antitumor functions of AD T cells in vitro and in vivo. Exogenous fumarate supplementation or healthy AD mitochondria transfer functionally mimicked the AD/APP phenotype in aging T cells, enhancing their antitumor activity to control tumor growth. Moreover, T cells isolated from aging donors showed elevated mitophagy with fumarate depletion, which was restored in T cells isolated from age-matched patients with AD. Together, these findings show that AD protects T cells against ceramide-dependent mitophagy and fumarate depletion to enhance antitumor functions.
The reduced cancer risk in Alzheimer’s disease patients is mediated by the amyloid-β 40 peptide, which inhibits aging-dependent mitophagy in T cells to improve antitumor immunity.
Comment on the article: Ceramide Links Alzheimer’s Disease to Decreased Cancer Risk: A Lipid Behind the Enigma of Inverse Comorbidity
For decades, epidemiologic studies have consistently reported an inverse comorbidity between Alzheimer’s disease (AD) and cancer: Individuals with AD are less likely to develop cancer and vice versa. The biological basis of this paradox has remained largely unresolved. A study by Kassir and colleagues in this issue of Cancer Research provides a compelling mechanistic insight into this paradox by demonstrating that amyloid precursor protein and its cleavage product Aβ40, known for their pathologic accumulation in the AD brain, also accumulate in peripheral T cells where they suppress mitochondrial ceramide production and lethal autophagy. This preservation of mitochondrial function enhances the antitumor immunity of T cells. Previous work has established that ceramide can promote neurodegeneration in the brain. The suppression of ceramide generation by amyloid precursor protein and Aβ40 in the periphery, thereby preserving mitochondrial integrity and supporting anticancer immunity, further establishes ceramide as a context-dependent regulator of cell fate and a key factor in the inverse AD–cancer relationship.
News article commenting on the academic paper: Notorious Alzheimer’s protein also supercharges cells against cancer
“What we found is that the same amyloid peptide that is harmful for neurons in Alzheimer’s is actually beneficial for T cells in the immune system,” said Besim Ogretmen, PhD, associate director of Basic Science at Hollings, and the study’s corresponding author. “It rejuvenates the T cells, making them more protective against tumors.”
“When you deplete fumarate, you increase mitophagy much more,” Ogretmen said. “Fumarate no longer binds proteins involved in that process, so the proteins become more active and induce more mitophagy. It’s like a reinforcing feedback loop.”
T cells from older humans showed the same pattern: too much mitophagy and low fumarate. But in T cells from older people with Alzheimer’s, these problems had been corrected, just as they had in mouse models.
“Older T cells began functioning like young, active T cells again,” Ogretmen said. “That was an incredible finding because it suggests a whole new way to think about rejuvenating the immune system.”
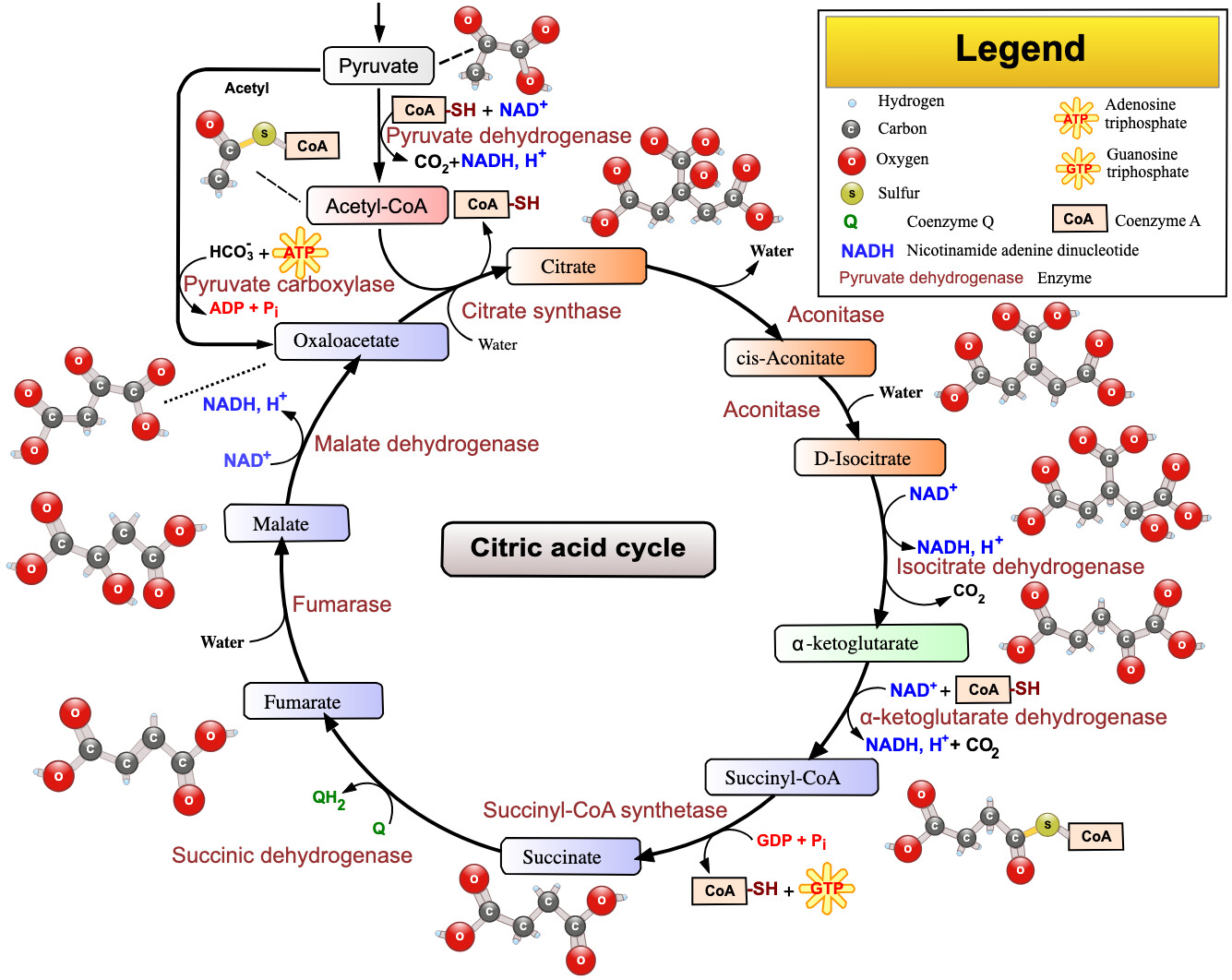
Fumarate in the citric acid cycle: