Talking with my Chemistry PHD friend about this issue of validating that the amino acid order in the peptide is correct, (and how to test for it) - he guided me towards these approaches, and suggested I’d need to work with the analytical chemistry lab (and get some support form them… these are not entirely standardized tests it seems).
1. Edman Degradation
Mechanism: Edman degradation sequentially removes and identifies one amino acid at a time from the N-terminus of a purified peptide. Under mildly alkaline conditions, the N-terminal amino group reacts with phenylisothiocyanate (PITC) to form a phenylthiocarbamyl (PTC) derivative. Subsequent treatment with an anhydrous acid, typically trifluoroacetic acid (TFA), cleaves the peptide bond of the N-terminal residue, yielding a thiazolinone derivative and leaving the rest of the peptide chain intact. The thiazolinone is extracted and converted into a more stable phenylthiohydantoin (PTH)-amino acid in aqueous acid, which is then identified via High-Performance Liquid Chromatography (HPLC) against known standards.
Strengths:
- Provides absolute sequence confirmation, which is highly precise for short peptides (up to 30–50 residues).
- Essential for verifying the exact N-terminal identity of recombinantly expressed proteins in biotech quality control.
Limitations:
- Inefficiency for large proteins: Yield drops with each cycle (typically 98% efficiency per cycle), leading to background noise that obscures the signal after ~50 cycles.
- N-terminal blockage: Completely fails if the N-terminus is chemically modified (e.g., formylation, acetylation, or pyroglutamate formation).
- Cannot identify positionally complex Post-Translational Modifications (PTMs) or cross-links efficiently.
2. Mass Spectrometry (MS)
Mechanism: Mass spectrometry has superseded Edman degradation for high-throughput proteomics. It determines sequences by measuring the mass-to-charge ratio () of ionized peptides and their fragments in a gas phase.
Dominance in Biotech:
- High Sensitivity: Requires only femtomolar to attomolar amounts of sample.
- Throughput & Complexity: Capable of analyzing complex mixtures (e.g., whole-cell lysates) without prior extensive purification.
- PTM Identification: The gold standard for mapping PTMs (phosphorylation, acetylation, advanced glycation end-products), which are critical variables in aging and longevity research.
Standard “Bottom-Up” Workflow:
- Proteolytic Digestion: Cleave the target protein into manageable peptides using a specific protease (e.g., Trypsin, which cleaves strictly C-terminal to Arg and Lys).
- Chromatographic Separation: Separate the complex peptide mixture via liquid chromatography (LC) directly coupled to the mass spectrometer.
- Ionization: Volatilize and ionize peptides using Electrospray Ionization (ESI) or Matrix-Assisted Laser Desorption/Ionization (MALDI).
- Tandem MS (MS/MS) Fragmentation: Isolate specific peptide precursor ions and fragment them using Collision-Induced Dissociation (CID) or Higher-energy Collisional Dissociation (HCD). This primarily breaks the peptide bonds, generating sequence-specific -ions (N-terminal fragments) and -ions (C-terminal fragments).
- Bioinformatic Reconstruction: Match the experimental MS/MS spectra against theoretical spectra generated from protein databases, or use computationally demanding de novo sequencing algorithms when a reference genome is unavailable.
3. Indirect Sequencing via DNA/RNA Sequencing
Mechanism: Deriving the primary structure of a protein by sequencing its corresponding gene or mRNA transcript and utilizing the standard genetic code to computationally translate the nucleotide sequence.
Caveats in Applied Biology:
- The “Static” Genome Problem: Fails to capture the dynamic reality of the proteome. It cannot detect alternative splicing variants unless RNA-seq is specifically utilized and properly mapped.
- Blind to Maturation: Cannot detect vital proteolytic processing events (e.g., cleavage of signal peptides or pro-hormones).
- Blind to PTMs: Offers no data on the ~300 known biological modifications that regulate protein function, stability, and half-life—data essential for developing structural analogs in biopharmaceutical engineering.
4. Hybrid Approaches & The Research Frontier
Modern proteomics relies on multi-omic integration. Combining genetic database searching (genomics/transcriptomics) with high-resolution MS/MS allows for rapid mapping of massive biological systems.
Scholarly Debates & Emerging Frontiers:
A current limitation in the field is the reliance on “bottom-up” MS, which destroys macroscopic protein context. The exact combination of PTMs on a single unbroken protein molecule (its “proteoform”) is lost.
- Top-Down Proteomics: Involves ionizing and fragmenting intact proteins. It maps the true proteoform but is severely limited by decreasing ionization efficiency and complex spectral overlaps as protein mass increases.
- Single-Molecule Protein Sequencing: Analogous to nanopore DNA sequencing, research is currently active in designing nanopores or using fluorophore-labeled Edman-like degradation to read individual protein molecules. This remains a significant technical challenge requiring highly engineered biological or solid-state pores.
Summary Table: Analytical Profiling of Peptides
| Method | Primary Utility | Major Limitations |
|---|---|---|
| Edman Degradation | Absolute N-terminal sequence confirmation of purified short peptides. | Blocked by N-terminal modifications; severe yield drop-off for long chains (>50 residues). |
| Mass Spectrometry (MS) | High-throughput, highly sensitive sequencing; PTM mapping; complex mixture analysis. | Relies heavily on computational reconstruction and databases; susceptible to ion suppression artifacts. |
| Amino Acid Analysis | Accurate quantitation of total amino acid ratios; determining absolute protein concentration. | Provides no sequence order; specific residues (Trp, Asn, Gln, Cys) are damaged or altered during standard hydrolysis. |
| Genetic Sequencing | Rapid, high-accuracy prediction of theoretical primary structure. | Completely ignores translational processing, proteolysis, and all post-translational modifications. |
Would you like me to detail the specific bioinformatic algorithms and statistical scoring models (e.g., SEQUEST, Mascot, or deep learning tools) used for de novo MS/MS sequence reconstruction?
I heard back from Finnrick on my questions about what they test, and don’t test yet:
Thanks for reaching out and for sharing these important concerns from your community!
Here’s how we currently cover the areas you mentioned:
Endotoxins (LPS): Yes, we offer endotoxin testing as a paid add-on.
Heavy Metals: Yes, we provide heavy metals testing, including a basic FDA 4-panel and more extensive panels on request.
Trifluoroacetic Acid (TFA) and other solvents: We offer solvents testing as a paid add-on.
Dimerization and Aggregation: Our standard peptide testing focuses on identity, purity, and quantity via HPLC, which may not fully detect subtle peptide aggregation or dimerization. We’re exploring expanding capabilities here.
Amino Acid Sequence Accuracy: Our identity testing confirms the peptide’s presence but cannot distinguish scrambled sequences that elute similarly in LC. This is a known challenge industry-wide.
Regarding the “reagent” test you mentioned, that typically refers to our standard panel covering identity, purity, and quantity.
We’re actively working to expand testing options based on feedback like yours, so please keep the suggestions coming!
You can find more details and submit samples here: Free Peptide Testing + Optional Endotoxin and Heavy Metals Analysis | Finnrick
Let me know if you’d like to discuss further or have any other questions!
Best,
Caanan
Note that finnrick is not a lab, more like a lab broker. You need to ask them which labs perform the tests you’re interested in. Lab quality in general varies widely.
Yes - but it’s really hard to tell lab quality. It would seem that Finnrick is the source (purchaser) for a large number of tests, so they would likely have the best idea of quality of the tests (but do they do comparisons - using same sample between multiple labs to check reliability)?
Generally the companies (analytical chemistry labs) who do the most of a given type of test, are going to be the ones that can do it best, and in the most reliable manner. Like anything, the more you do something, the more you’ve standardized the process, and the better the consistency is.
Given this, it would seem that the labs that Finnrick uses, are going to be one of the better labs for doing this, just due to the volume. But perhaps there are other labs that do most of the industry testing in the USA? Does anyone know the top Peptide analytical chemistry labs? I’ll try Google Gemini.
Finnrick wouldn’t know lab quality unless they did comparisons between them; they just assume all labs are created equal. Anyway, this is one of the many reasons most peptide communities don’t trust this company and its ratings.
Here is the list of labs they use :
Mz biolabs
Chromate
Janoshik
BT Labs
TrustPointe
Krause Analytical
The only recommendation I can make is for Janoshik. By volume, they probably do 5-10x of the tests that the rest of the labs in this list do combined. By reputation, they are considered the gold standard.
TrustPointe is also solid, although there are some peptides they cannot test, check with them.
I recommend you stay away from BT and Krause.
Agree that Janoshik is the gold standard. Trustpointe also a solid option for the services they offer, and they’re in the USA.
Ok, but how does Finnick manage to do the testing for free then. My understanding is that they offer free testing (basic) you only pay for shipping? Wouldn’t they have to pay a fee (however small) should they use a third party.
On the pro side, perhaps we can get some peptides (though probably not SS-31) from compounding pharmacies in the US (which would seem potentially safer than ordering from some random seller in China). But… we still don’t have any good human data / clinical trials on most of these:

I find it interesting how the news coverage of something like peptide use (research grade) starts in one small report, then ripples out across the globe.
I must be the only one using peptides, but not using bpc. These stories about peptides are pretty much variations of each other. It’s always the same peptides being named.
This was the first time I’d seen a positive commentary on this peptide… though I don’t look around on this topic much:
Katie’s been injecting GHK-Cu, a copper peptide, for several weeks now and she’s confident it’s making a difference to her skin. So much so, she says, the stretch marks she developed after having her two children have almost disappeared.
I have a lot of women who swear by GHK-cu, it’s become one of the more popular ones. They have all come from 1 client who told 2 friends who told 2 friends kind of thing.
Even though I’ve been formulating our topical product with GHK-cu for 4 years now and have many repeat customers for that, I was a bit skeptical using it as an injectable. After it started to go a bit viral among my little group, my wife and I started using it about 3 months ago.
While we both have “good skin” I’ve had more compliments on my complexion in the past month than in my entire life time LoL!
I’ve noticed a significant reduction in fine wrinkles, the little ones under the eye are all gone. When I saw that I was sold. Also as we age the number of large pores increases, I don’t seem to have many large pores.
The media loves to spread fear porn. Looks like they moved on from trying to convince the masses that melatonin causes heart failure (LOL)
I’ve seen a lot of great reports, but I have not taken the plunge because I worry about injecting too much copper. If I harm my health, I want to be able to say I did it to make myself healthier, not for vanity. I’m would guess if you run labs often, it wouldn’t be a worry?
I’ve heard it stings, too… but people mix it with other stuff to minimize the ‘bite’.
I don’t know how much copper risk there is if you inject it, anyone?
@Steve_Combi will you share your thoughts?
I am about to order the powder to mix with serums as a topical.
EDIT:
Perplexity
https://www.perplexity.ai/search/toxicity-risk-of-ghk-cu-inject-aJz8raWoR5mh3bx.RyOy_A
Copper risk depends on whether you have Wilson’s disease (very high risk) or are a carrier for it (lower risk). Anyone considering GHK-cu injection should have copper serum tested before a cycle, early in a cycle, and likely throughout and at the end of a cycle. Particularly their first cycle. They may also consider relatively low accuracy genetic testing for Wilson’s.
Personally I found the impacts of injected GHK-cu marginal at best. I suspect many folks would find better results with basically cleaning and moisturizing on their skin, and simply the passage of time for issues like stretch marks after significant weight loss.
Topical application has some meaningful research supporting it, though the impacts again are not dramatic. Injected application has nearly no research supporting it for significant impact.
In spite of a dozen vials sitting in my freezer, I won’t be cycling this one again.
Frankly, similar story for BPC and TB4. The benefits are hard to detect even during injury recovery. While the marginal risk (mechanistic viability of cancer risk without significant data showing that impact) is a low benefit to risk margin in my book.
Cycled “Glow” three times and just concluded what will be my last cycle. I’ll keep the BPC and TB4 around in case I get bored and want to try again after future injuries. But the GHK-cu will just be made into terribly expensive skin lotion and serum.
That said, I’ll continue Reta and Tesa/Ipa from these vials from China.
I’ve also used GHK-cu both topically and subcutaneously. Frankly, the results are ambiguous at best. Now that I’ve stopped using it, my mirror image may show signs of accelerated aging. If it does, that’s fine with me. I don’t care anymore.
I’ve had better results with TB-4. And pain relief I do care about. It is not like morphine, but it is more effective than ibuprofen or acetaminophen, providing both immediate and long-term improvement.
Retatrutide remains the uncrowned king of gray market peptides.
What do people think will happen with the peptide market this year?
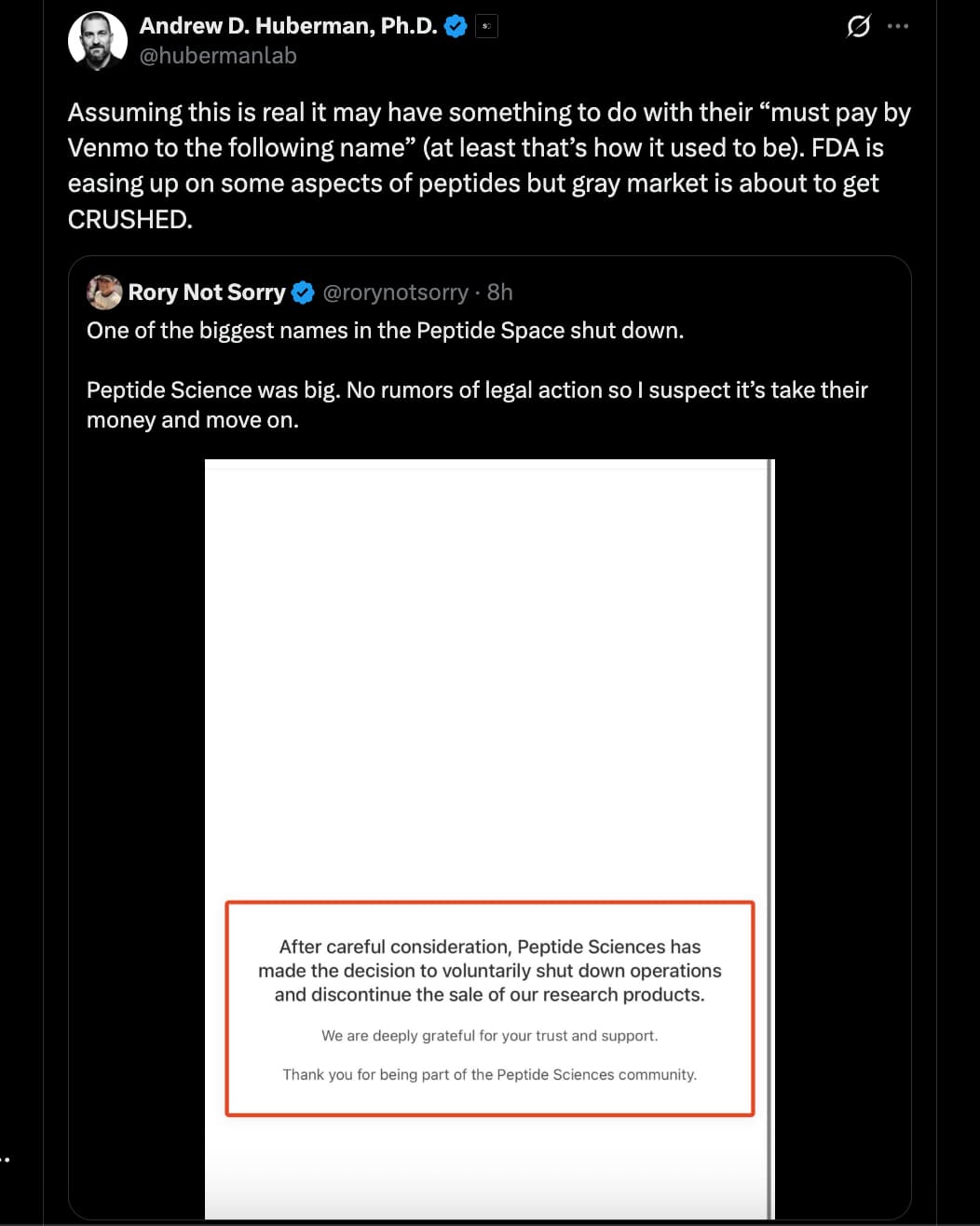
Source: https://x.com/hubermanlab/status/2030012241643458857?s=20
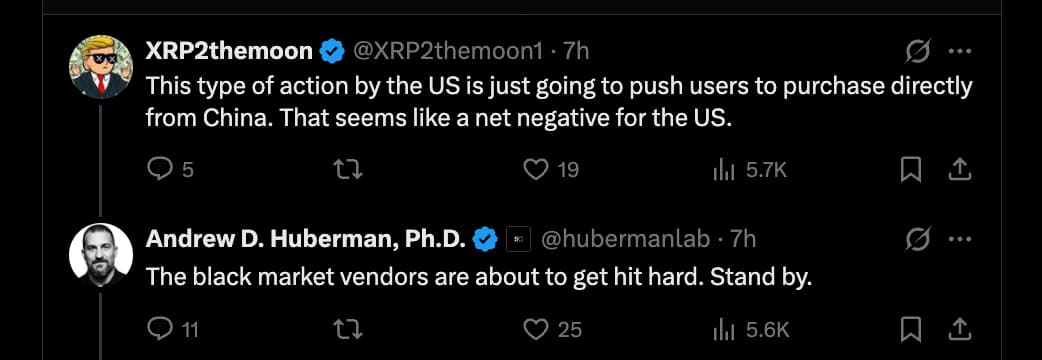
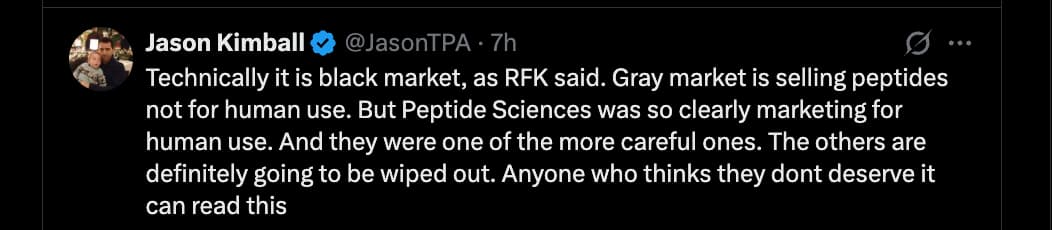
You’ll have to specify which peptide market. The one I buy from seems fine so far. Many vendors are still recovering from the holidays, and have either just started shipping orders, or manufacturing new batches.
As for the possible reason Huberman hinted at, yes, losing your payment processors is a big blow. I’m sure there are other reasons they shut down, as walking away from a business rumored to make over 5M/month is not something people decide overnight.
Maybe they heard about this guy, who is being federally prosecuted for selling gray markets peps: