Study Design Specifications
-
Type: Human genetic association study (Observational multi-cohort).
-
Subjects: 1.1 million adult human participants from 11 cohorts (including the UK Biobank, Geisinger Health System, and others).
-
Sub-cohort: 13,454 individuals with rare (allele frequency < 0.5%) nonsynonymous variants in the MSTNgene.
-
Imaging validation: 77,572 individuals assessed via whole-body Dixon MRI.
Mechanistic Deep Dive
Myostatin (GDF8) is a TGF-beta family member that restricts muscle growth by binding to ActRII/ALK-4 or 5 receptors, which in turn suppresses downstream hypertrophic pathways such as Akt/mTORC1. The genetic variants identified in this study induce muscle hypertrophy primarily by compromising the bioavailability or structural integrity of the mature myostatin ligand.
-
Protein Dynamics: Mutations such as Arg65His and Thr115Met, located in the prodomain, likely hyper-stabilize the inactive latent complex, preventing the proteolytic cleavage necessary to release the active growth factor. Conversely, the Arg283Cys mutation in the mature growth factor domain creates an unpaired cysteine, likely promoting protein aggregation and degradation before it can bind its receptor. [Confidence: High].
-
Tissue-Specific Priorities: The data strongly suggest that the musculoskeletal system is the primary responder to systemic myostatin reduction, with secondary reciprocal reductions in adipose tissue volume. The reduction in adiposity may be a secondary metabolic consequence of increased skeletal muscle energy expenditure. [Confidence: Medium].
Novelty
Prior human genetic evaluations of MSTN lacked the statistical power to definitively detect continuous, dose-dependent phenotypic effects of heterozygous function-disrupting variants. This study provides the first definitive, large-scale human genetic evidence that lifelong myostatin inhibition safely enhances muscle mass and strength while decreasing fat mass, without inducing adverse cardiovascular (e.g., hypertrophic cardiomyopathy) or reproductive (e.g., altered FSH levels, PCOS) side effects. [Confidence: High].
The analysis of biological and medical claims extracted from the provided text reveals a mix of robust clinical validation and early-stage pre-clinical data. Maintaining skeletal muscle mass is a critical factor in extending healthspan, making the rigorous verification of myostatin-inhibition mechanisms essential for developing actionable longevity protocols. Below is the objective hierarchy of evidence for each specific claim.
Claim 1: Homozygous loss-of-function mutations in the myostatin (MSTN) gene cause gross skeletal muscle hypertrophy in humans.
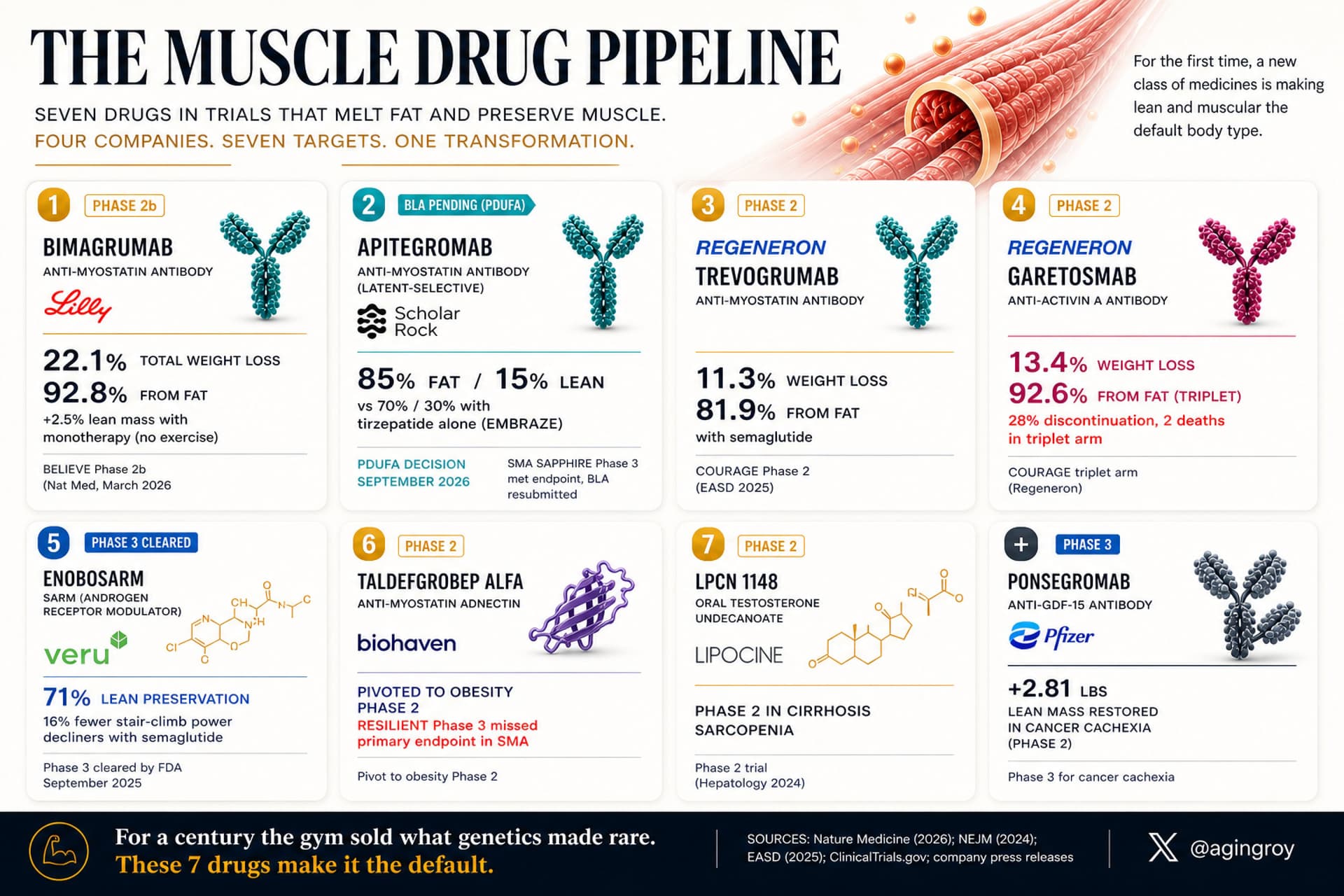
Claim 2: Pharmacological blockade of ActRII/myostatin pathways (via Bimagrumab) increases lean muscle mass while significantly reducing fat mass in adult humans.
The Strategic FAQ
1. Question: Does the hyper-muscularity from MSTN variants translate to functional tendon and ligament strength, or just contractile tissue volume? Answer: The paper notes a highly significant increase in handgrip strength correlating with the variants, proving the contractile tissue is functional. However, the study does not directly measure tendon/ligament tensile strength or collagen synthesis, leaving a minor gap regarding injury risk under heavy mechanical load.
2. Question: Given that the data reflects lifelong, developmental gene suppression, how accurately does this predict the effect of acute pharmacological blockade in a 60-year-old? Answer: Lifelong genetic suppression allows for developmental compensation, which is a known translational limitation. However, concurrent clinical trials with Bimagrumab in older adults confirm that acute blockade does replicate the genetic phenotype (lean mass gain, fat mass loss), validating the genetic blueprint.
3. Question: Does ActRII/myostatin blockade accelerate the depletion of satellite cell pools, leading to premature muscle senescence? Answer: The genetic data does not indicate premature sarcopenia in older carriers. The paper identifies a homozygous carrier over 65 years old who maintained extreme hypertrophy (+37.7% Sarcopenia Index), suggesting satellite cell exhaustion is not a near-term clinical barrier.
4. Question: Did the genetic analysis reveal any unintended compensatory up-regulation of other TGF-beta family members, such as Activin A? Answer: The paper explicitly notes that Activin A (INHBA) is highly conserved and intolerant to loss-of-function, but it does not measure compensatory serum up-regulation in MSTN variant carriers. Because ActRII receptors bind multiple ligands, compensatory up-regulation is highly probable in a clinical setting.
5. Question: The study relies heavily on BIA (bioelectrical impedance) for the full cohort. How badly does hydration status skew the fat-free mass data? Answer: BIA is notoriously sensitive to hydration and glycogen status. The authors aggressively mitigated this by using an MRI sub-cohort (n=77,572) to validate the BIA data. The MRI confirmed actual muscle water volume increased, proving the BIA signal was a true morphological change, not a hydration artifact.
6. Question: Is there a threshold where myostatin inhibition begins to induce cardiac hypertrophy? Answer: No. The study explicitly investigated left ventricular myocardial wall thickness using cardiac MRI and found no association with increased wall thickness. Counterintuitively, there was a nominal association with lower wall thickness, clearing a major pre-clinical safety hurdle.
7. Question: Can myostatin inhibition be achieved via oral small molecules, or are we restricted to injectables? **Answer:**Currently, we are restricted to injectable biologics (mAbs). Oral small molecules have routinely failed due to the structural complexity of the ActRII receptor and the need for high target specificity without off-target kinase inhibition.
8. Question: How does the reciprocal fat loss actually occur if myostatin specifically targets muscle? Answer: The exact mechanism is debated, but it is primarily a secondary metabolic consequence. Increased skeletal muscle mass dramatically increases resting energy expenditure and acts as a massive glucose/lipid sink, indirectly starving adipose tissue and driving lipolysis.
9. Question: Were there any negative effects on bone mineral density (BMD) since TGF-beta signaling is heavily involved in bone remodeling? Answer: The study does not explicitly report on BMD. However, ActRII blockade in separate clinical trials generally shows neutral or mildly positive effects on bone density, likely driven by the increased mechanical loading of the larger muscle mass on the skeletal structure.
10. Question: What happens when you stop an ActRII inhibitor? Do you lose the muscle immediately? Answer: Clinical trial data suggests the muscle hypertrophy is relatively stable for several months post-cessation, but eventually regresses to a new homeostatic baseline dictated by the individual’s diet, exercise, and age.