Part 3: Claims & Verification
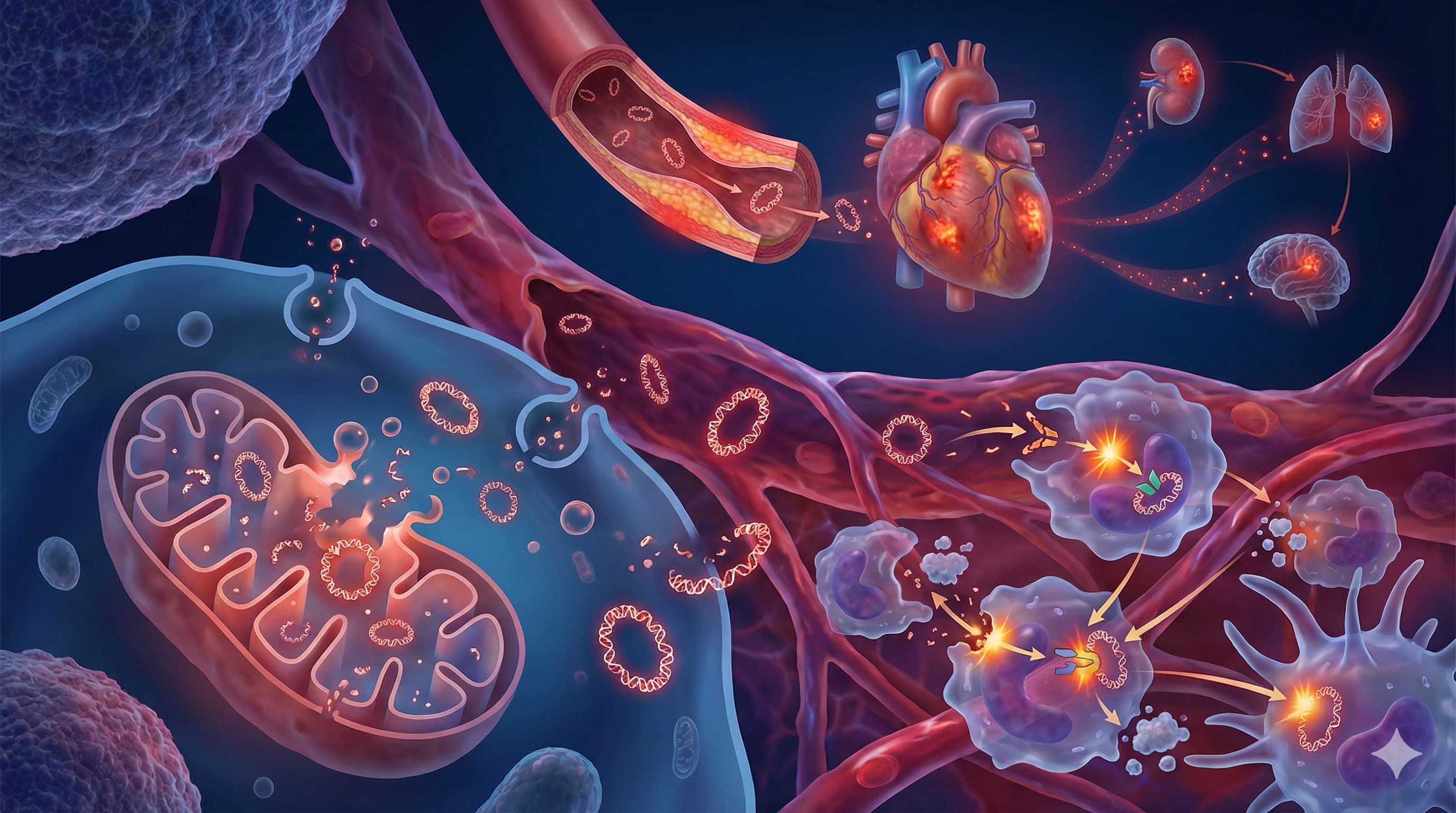
Claim 1: Cytosolic leakage of mitochondrial DNA (mtDNA) acts as a Damage-Associated Molecular Pattern (DAMP), activating the cGAS-STING pathway to trigger type I interferon release and sterile inflammation.
-
Evidence Level: Level A (Systematic Reviews of primarily in vitro/in vivo data) & Level D (Pre-clinical).
-
External Verification: Live search confirms this is a foundational consensus in innate immunology and tumor biology. Molecular Mechanisms of mtDNA-Mediated Inflammation (2021).
-
Translational Gap: HEAVILY FLAGGED. While the cGAS-STING pathway operates in humans, therapeutic interventions proposing to block this pathway to prevent cardiovascular aging are currently confined to Level D pre-clinical models (mice/cell lines). Systemic cGAS-STING inhibition in humans poses severe risks to antiviral and anti-tumor immune surveillance. Actionable human longevity protocols targeting this specific node safely do not yet exist.
Claim 2: Circulating cell-free mtDNA copy number (ccf-mtDNA-CN) is a viable diagnostic and prognostic biomarker for cardiovascular disease risk, severity, and atherosclerotic progression.
-
Evidence Level: Level C (Human Observational / Cohort Studies) & Level A (Scientific Statements).
-
External Verification: Validated by major cardiovascular authorities. Clinical cohorts consistently demonstrate that elevated ccf-mtDNA correlates with adverse cardiovascular outcomes and systemic inflammation. Mitochondrial Genetics in Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association (2025).
-
Knowledge Gap: As a biomarker, ccf-mtDNA lacks assay standardization across clinical settings, making baseline comparisons difficult. Furthermore, correlation does not equal causation; scholarly debate remains regarding whether elevated ccf-mtDNA is a primary driver of human cardiovascular events or merely a secondary byproduct of cellular necrosis and tissue damage.
Claim 3: Oxidized mtDNA directly primes and activates the NLRP3 inflammasome, leading to caspase-1 activation, IL-1beta secretion, and pyroptotic cell death, which accelerates atherosclerosis.
-
Evidence Level: Level A (Systematic Reviews) & Level D (Pre-clinical).
-
External Verification: Live search corroborates the mechanistic link between oxidized mtDNA and NLRP3 activation, specifically in macrophage foam cells within atherosclerotic plaques. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis (2018) and Mitochondrial DNA in atherosclerosis research progress: a mini review (2025).
-
Translational Gap: Mechanistic proof is largely derived from knockout mice (e.g., NLRP3-/- models). While general NLRP3 inhibition (e.g., via low-dose colchicine) has shown clinical benefit in human cardiovascular disease (Level B RCTs), the specific quantitative contribution of mtDNA-driven NLRP3 activation versus other triggers (e.g., cholesterol crystals) in human atherogenesis remains unquantified.
Claim 4: Pharmacological or lifestyle activation of autophagy/mitophagy (via compounds like rapamycin or spermidine) enhances the clearance of damaged mitochondria, preventing mtDNA leakage and attenuating systemic inflammaging.
-
Evidence Level: Level D (Pre-clinical: Animal/In vitro) & Level E (Expert Opinion/Extrapolation).
-
External Verification: The biochemical mechanism of mitophagy clearing defective organelles to prevent DAMP release is thoroughly documented in cellular and murine models. Mitochondrial quality surveillance: mitophagy in cardiovascular health and disease (2021).
-
Translational Gap: HEAVILY FLAGGED. This represents the primary actionable claim for longevity interventions, yet it relies entirely on Level D evidence. While compounds like rapamycin and spermidine successfully induce mitophagy and extend lifespan in mice and lower organisms, Level B evidence (Human RCTs) proving that these compounds prevent human cardiovascular aging specifically by reducing mtDNA leakage does not exist. This is currently informed speculation requiring long-term human outcome data.
Claim 5: Endosomal Toll-like receptor 9 (TLR9) recognizes unmethylated CpG motifs in escaped mtDNA, driving NF-kB-dependent pro-inflammatory cascades.
-
Evidence Level: Level A (Reviews) & Level D (Pre-clinical).
-
External Verification: Verified. The structural similarity between bacterial DNA and mitochondrial DNA (both containing hypomethylated CpG motifs) makes mtDNA a potent TLR9 agonist when internalized by phagocytic cells. Mitochondrial DNA Dysfunction in Cardiovascular Diseases: A Novel Therapeutic Target (2025).
-
Translational Gap: In vivo validation of TLR9-mediated cardiovascular tissue damage is predominantly established in murine knockout models rather than human interventional trials.
The Strategic FAQ
1. Does artificially clearing ccf-mtDNA directly extend maximum lifespan, or is it just a downstream marker of existing damage? Unproven in humans. While pre-clinical models show that upregulating DNases (which clear extracellular DNA) reduces inflammation, there is no direct evidence that selectively clearing ccf-mtDNA alone extends human lifespan. It is likely both a marker of bioenergetic failure and a secondary amplifier of the aging process.
2. If we inhibit the cGAS-STING or TLR9 pathways to stop “inflammaging,” don’t we severely compromise the immune system? Yes. This is the primary translational bottleneck of the paper’s proposed therapeutic targets. Systemic inhibition of cGAS-STING or TLR9 in humans presents an unacceptable risk for opportunistic viral infections and suppresses anti-tumor immune surveillance. Safely targeting this axis requires extreme tissue specificity.
3. Can longevity clinics accurately measure ccf-mtDNA in standard blood panels right now? No. Assay standardization does not exist. The centrifugation speed, freezing protocols, and PCR primers wildly alter the readouts of ccf-mtDNA. Distinguishing between baseline physiological mtDNA turnover and pathological leakage is currently unreliable outside of controlled academic research.
4. The paper notes sex differences in mtDNA signaling. How does this impact longevity protocols? Males exhibit higher circulating mtDNA levels and significantly stronger TLR9-mediated inflammatory responses (greater IL-6/TNF-α release). Females generally show blunted TLR9 responses. This suggests that men may derive a proportionally larger cardioprotective benefit from interventions that clear mtDNA or dampen sterile inflammation.
5. How does physical exercise compare to pharmacological mitophagy activators for clearing damaged mitochondria? Exercise is currently the most potent, validated, and systemic activator of mitochondrial quality control and mitophagy. Vigorous exercise temporarily increases localized stress, which robustly upregulates the clearance of defective mitochondria before their membranes become permeable, effectively cutting off the mtDNA leak at the source.
6. Do interventions targeting the NLRP3 inflammasome block this specific aging vector? Yes, but further downstream. Colchicine, an FDA-approved drug that inhibits NLRP3, has shown significant cardiovascular benefit in trials like LoDoCo2. It intercepts the inflammatory cascade after mtDNA has leaked and oxidized, reducing pyroptosis and plaque instability, though it does not prevent the initial mitochondrial damage.
7. Is there a role for exogenous NAD+ precursors (NMN/NR) in mitigating this pathway? Indirectly, yes. NAD+ activates sirtuins (specifically SIRT1 and SIRT3), which are master regulators of mitochondrial biogenesis and mitophagy (via PINK1/Parkin pathways). Maintaining high NAD+ pools theoretically accelerates the clearance of defective mitochondria, preventing the accumulation of the BAX/BAK pores that release mtDNA.
8. What is the primary cellular source of the circulating mtDNA driving cardiovascular disease? The data points to dying cardiomyocytes and stressed endothelial cells as primary sources during ischemic events. Additionally, Neutrophil Extracellular Traps (NETs)—webs of DNA expelled by immune cells—contain mtDNA that locally amplifies inflammation in failing cardiovascular tissue.
9. Can we just use Metformin to block this, as suggested in the paper? Metformin is noted in the literature to normalize mitochondrial function and delay senescence via the Mfn2-cGAS pathway. However, its exact efficacy in preventing mtDNA release specifically in human cardiovascular aging remains entangled with its broader AMPK-activating and glucose-lowering effects.
10. Why is autophagy prioritized over direct DNA-degrading enzymes (DNases) for therapy? Autophagy (mitophagy) is an intracellular, upstream preventative mechanism. It degrades the entire faulty mitochondrion before the mtDNA can escape into the cytosol or bloodstream. DNases only work extracellularly, meaning they only clean up the mess after the cell has already ruptured or initiated a severe inflammatory response.