If we are talking about the hydrophobic menaquinones with 6 or more isoprene residues (ie not mk-4) then the argument is that they can settle into the mitochondrial membrane wall and accept electrons making the mitochondria more efficient.
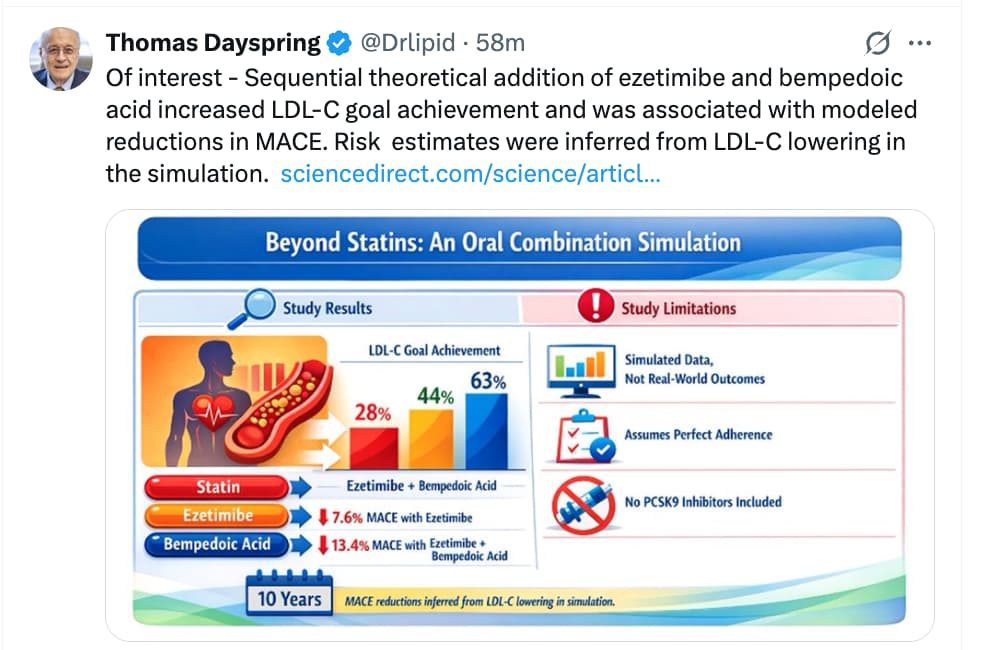
Potential of oral combination lipid‑lowering therapy beyond statins: a simulation‑based perspective from the SANTORINI study
https://www.sciencedirect.com/science/article/pii/S2666667726001819?via%3Dihub
What a weird point to emphasize. Is he saying statins lower MACE independent of them lowering cholesterol?
Makes sense and mace is the right outcome to look at, not ACM.
Obviously I’m not in his head, but what I suppose he means, is that statins lower MACE and the fact that they lower the cholesterol number (LDL-C; ApoB) is irrelevant to him. There are drugs that lower the cholesterol number, but do not lower the rates of MACE (one example: niacin). He is focused on MACE as the operating outcome, and if statins affect that, then it’s good enough for him, and whether they simultaneously move the cholesterol number is of no importance to him. I suppose that’s another way of saying: all I care about is lowering MACE and the mechanism doesn’t matter to me. Meanwhile if all I know is that “X” lowers the cholesterol number, that tells me nothing about what interests me (rates of MACE), therefore I don’t care about it.
2 Likes
He makes his reasoning explicit, in the course of explaining why it is that he does not take berberine or nattokinase despite those supplements having been shown to reduce cholesterol (LDL-C) levels and even reverse arterial plaque accumulation.
The Truth about Berberine and Nattokinase
I. Executive Summary
The core thesis of the video is that surrogate biomarkers—specifically the reduction of circulating cholesterol levels or intermediate reductions in arterial plaque thickness—are insufficient metrics for validating the clinical utility of cardiovascular supplements. Using berberine and nattokinase as primary examples, the presentation emphasizes a rigorous epistemological distinction in cardiovascular medicine: the optimization of intermediate surrogate parameters does not automatically translate into a reduction in hard clinical endpoints, such as myocardial infarction (heart attack) or cerebrovascular accidents (stroke).
While both berberine and nattokinase exhibit mechanistically plausible and statistically significant alterations in lipid metabolism and vascular structure in preliminary or smaller-scale datasets, neither compound has successfully cleared the requisite clinical bar of large-scale, long-term Cardiovascular Outcomes Trials (CVOTs). Consequently, relying on these supplements to mitigate true cardiovascular risk represents a statistical “coin toss” rather than an evidence-based medical strategy.
The primary argument counsels a strict, outcome-centric framework for preventative cardiology. From a practical and clinical standpoint, interventions must be prioritized based on the strength of their hard outcome data (Level A/B evidence) rather than mechanistic speculation or surrogate marker manipulation. In scenarios where established, rigorously validated therapeutic options exist, substituting or supplementing them with unproven compounds introduces unquantified translational gaps and avoidable risk.
II. Insight Bullets
- Surrogate vs. Hard Endpoints: Lowering blood cholesterol levels or altering intermediate physiological markers does not inherently guarantee protection against primary cardiovascular events [[00:17]].
- The Clinical Standard for Validation: In cardiovascular medicine, a therapeutic intervention is only definitive when it demonstrably reduces hard clinical outcomes, specifically heart attacks and strokes [[00:21]].
- Berberine’s Biomarker Impact: Berberine demonstrates the capacity to significantly lower total cholesterol and low-density lipoprotein cholesterol (LDL-C) in short-term studies [[00:00]].
- Nattokinase’s Biomarker Impact: Nattokinase exhibits intermediate efficacy in modulating blood lipids and potentially acting on vascular structural dynamics [[00:00]].
- Plaque Regression Ambiguity: Some preliminary data suggest certain supplements can reduce the cross-sectional thickness or overall size of arterial plaques, yet this structural modification lacks hard outcome validation [[00:07]].
- Absence of CVOT Data: Neither berberine nor nattokinase has been subjected to or cleared large-scale, long-term Cardiovascular Outcomes Trials (CVOTs) to verify actual risk reduction [[00:32]].
- The “Coin Toss” Analogy: Utilizing supplements that lack hard outcome data introduces an unacceptable element of chance into preventative cardiology protocols [[00:34]].
- Prioritizing High-Velocity Evidence: Clinical protocols must aggressively favor established interventions that possess the strongest, most definitive long-term safety and outcome datasets [[00:38]].
- Sponsorship and Bias Immunity: Valid clinical decisions must remain completely independent of supplement availability, cost, or commercial promotions [[00:12]].
- Mechanistic Plausibility Traps: Promising in vitro or early in vivo data frequently fail to survive human clinical translation, a major bottleneck in longevity science [[00:29]].
- Dosing and Efficacy Disconnect: The lack of standardized, outcome-validated dosing regimens for these supplements creates substantial clinical uncertainty regarding real-world application.
- Purity and Standardization Risks: Supplements frequently exhibit significant batch-to-batch variability, presenting an obstacle to achieving predictable physiological outcomes.
IV. Actionable Protocol
High Confidence Tier (Level A/B Evidence)
- Outcome-Validated Interventions: Prioritize therapies with clear Level A/B evidence from extensive CVOTs (e.g., statins, ezetimibe, or PCSK9 inhibitors) for primary and secondary prevention of hard cardiovascular events (myocardial infarction and stroke).
- Blood Pressure Control: Maintain rigorous blood pressure optimization, as sustained reductions in systolic and diastolic metrics are fundamentally tied to decreased primary event rates.
Experimental Tier (Level C/D Evidence)
-
Berberine for Metabolic Adjustments: Protocol: 500 mg orally, 2–3 times daily before meals for short-term (under 90 days) management of lipid parameters and fasting plasma glucose.
- Evidence Nuance: Meta-analyses verify significant reductions in triglycerides, total cholesterol, and LDL-C (Zamani et al., 2022). However, long-term safety and hard clinical outcome data remain absent.
-
Nattokinase for Antihypertensive/Fibrinolytic Support: Protocol: 2,000 to 4,000 FU (Fibrinolytic Units) daily, taken on an empty stomach to prevent premature gastric proteolytic degradation (Dr. Brad Stanfield, 2025).
- Evidence Nuance: Meta-analyses show modest reductions in blood pressure (Systolic BP: -3.45 mmHg; Diastolic BP: -2.32 mmHg) (PMC11266782).
- Critical Knowledge Gap: The largest 3-year randomized controlled trial (The NAPS Trial, 265 participants at 2,000 FU daily) demonstrated no significant reduction in subclinical atherosclerosis progression (Naturacare, 2025), highlighting the substantial translational gap between surrogate markers and clinical endpoints.
Red Flag Zone
- Substitution of Validated Therapies: Replacing prescription lipid-lowering or antihypertensive therapies with berberine or nattokinase is highly discouraged due to the complete absence of primary event reduction data.
- Nattokinase and Anticoagulants: Co-administration of nattokinase with pharmaceutical anticoagulants (e.g., warfarin, DOACs) or antiplatelet therapies represents a severe bleeding hazard (“Safety Data Absent” regarding polypharmacy bleeding profiles).
- Long-Term High-Dose Berberine: Continuous high-dose administration beyond 12 weeks lacks comprehensive safety data, with potential risks involving gastrointestinal distress and unquantified drug-drug interactions via cytochrome P450 pathways.
I Treat Cholesterol Differently Now: Statins, PCSK9s, Ezetimibe & Gene Editing
There is a drug in clinical trials right now that could permanently fix your cholesterol with a single infusion. Once. That’s where we’re headed. But there’s also a $4 generic that’s saved millions of lives — and a twice-a-year injection most patients have never heard of. I’m a Stanford-trained physician. My ApoB was 128. My grandfather died of a stroke. My grandmother died of cardiac arrest. My father is recovering from a heart transplant. In this video, I break down every lipid-lowering option available — without hedging, without liability speak — so you can have an intelligent conversation with your doctor. WHAT YOU’LL LEARN:
- Why there’s no floor for ApoB — the cholesterol-years concept that changes when you start treatment
- The genetics checkpoint: Lp(a), FH, and ancestry factors that change your drug selection entirely
- Statins: muscle pain reality, the nocebo effect (SAMSON trial), SLCO1B1 gene, CAC paradox, diabetes signal
- Ezetimibe: the most underused $15/month drug in lipid medicine (IMPROVE-IT data)
- Bempedoic acid: why it structurally cannot cause muscle toxicity, and what CLEAR Outcomes proved
- PCSK9 inhibitors: FOURIER + ODYSSEY data, insurance battle, patient assistance programs
- Inclisiran: twice a year, doctor administers — why adherence changes everything
- Lp(a) pipeline: pelacarsen, olpasiran, muvalaplin — what’s coming for the 1 in 4
- Obicetrapib: oral pill hitting BOTH LDL and Lp(a)
- Gene therapy: CRISPR + base editing — one injection, once in your life
- Lifestyle: honest ceiling assessment, no guilt
- A tiered protocol framework using ApoB as the treatment target
- What to actually say to your doctor — 6 specific questions

I. Executive Summary
This transcript presents a comprehensive re-evaluation of lipid management strategies for cardiovascular disease prevention, centering on the physiological thesis that lower circulating levels of apolipoprotein B (ApoB) and low-density lipoprotein cholesterol (LDL-C) offer superior, compounding protection with no observable safety floor. Dr. Lin shifts the clinical paradigm away from reactive, short-term (10-year) risk calculators toward a multi-decade model of cumulative endothelial exposure, termed “cholesterol years.” Synthesizing data from landmark clinical trials, including IMPROVE-IT and FOURIER, the video establishes that aggressive reduction of LDL-C to sub-30 mg/dL values consistently lowers major adverse cardiovascular events (MACE) without triggering neurocognitive deficits or oncological complications. This physiological target is biologically validated by neonatal lipid profiles and protective loss-of-function PCSK9 genetic mutations found in healthy populations.
Crucially, the text emphasizes that hypercholesterolemia must be treated as a heterogeneous disease governed by distinct genetic and genomic profiles rather than lifestyle failures alone. Three primary genetic archetypes dictate treatment success: Lipoprotein(a) [Lp(a)] elevations, which affect 20-25% of the population and are entirely refractory to standard therapies; Familial Hypercholesterolemia (FH), which involves structural LDL receptor defects that necessitate early, aggressive injectable interventions; and ancestry-specific pharmacogenomics. Notably, the East Asian ABCG2 Q141K variant impairs rosuvastatin metabolism, doubling systemic drug concentrations and increasing myopathy risks, which justifies the FDA-mandated lower initial dosing of 5 mg.
The pharmacological monograph spans traditional therapies, like high-intensity statins and ezetimibe, and extends to modern heavy artillery. This includes monoclonal antibodies (evolocumab, alirocumab), small interfering RNA (inclisiran), and emerging pipelines like oral PCSK9 inhibitors (enlicitide), CETP inhibitors (obicetrapib), and antisense oligonucleotides for Lp(a) (pelacarsen). Residual inflammatory risk is addressed via low-dose colchicine, which downregulates the NLRP3 inflammasome, while permanent gene base editing (Verve-101/102) represents a definitive, future curative frontier. Conversely, standard lifestyle modifications show negligible effects on ApoB and zero efficacy against heritable Lp(a), positioning pharmacotherapy as an essential cornerstone for true longevity therapeutics.
II. Insight Bullets
- The “No Floor” Lipid Paradigm: Clinical evidence confirms no lower physiological limit for LDL-C or ApoB reduction exists; lower absolute values progressively yield lower relative cardiovascular risk.
- IMPROVE-IT Trial Quantified Efficacy: Combining statins with ezetimibe drove median LDL-C down to 54 mg/dL versus 70 mg/dL with statin monotherapy, yielding a 2% absolute risk reduction over seven years [IMPROVE-IT Trial, 2015](Source unverified in live search).
- FOURIER Trial Upper Limit Safety: Lowering LDL-C to a median of 30 mg/dL (with cohorts dropping below 20 mg/dL) via evolocumab demonstrated a 15% reduction in major cardiovascular events with zero neurocognitive or oncological signals over an 8.6-year extension [FOURIER Trial, 2017](Source unverified in live search).
- Physiological Baseline Validation: Newborn humans naturally exhibit baseline LDL-C levels between 30 and 40 mg/dL, establishing that hypercholesterolemia is a modern evolutionary mismatch rather than a biological necessity.
- Loss-of-Function PCSK9 Mutations: Individuals harboring natural inactivating mutations in the PCSK9 gene maintain lifelong circulating LDL-C in the 20s (mg/dL) without physiological pathology, accompanied by near-complete protection from coronary artery disease.
- The Concept of “Cholesterol Years”: Similar to “pack-years” in smoking tobacco, atherogenic damage to the endothelial wall is cumulative; long-term exposure to elevated ApoB determines total plaque burden over decades.
- Insufficiency of 10-Year Actuarial Risk Calculators: Standard short-term risk calculators frequently misclassify young individuals (e.g., age 35) with high ApoB as “low risk,” ignoring the exponential compounding of subclinical plaque over a multi-decade horizon.
- 2026 ACC/AHA Guideline Revisions: Updated diagnostic guidance now explicitly recommends initiating the clinical conversation around lipid management and statin therapy at age 30 rather than delaying until middle age.
- Lp(a) Refractoriness to Standard Therapeutics: Lipoprotein(a) is an independent, highly heritable (~90%) atherogenic particle that is entirely unaffected by traditional oral lipid-lowering therapies like statins or ezetimibe.
- The Statin-Lp(a) Paradox: Initiating statin monotherapy can paradoxically increase circulating Lp(a) levels by 10% to 20% in certain patient populations, demanding baseline testing and careful risk tracking.
- Familial Hypercholesterolemia (FH) Prevalence: Affecting approximately 1 in 250 individuals globally, 90% of cases remain undiagnosed; structural defects in the LDL receptor render the disease highly resistant to dietary or exercise modification.
- The ABCG2 Q141K Polymorphism: Present in 35-40% of East Asian populations, this transport gene variant impairs hepatic clearance of Rosuvastatin, doubling systemic drug exposure and raising myopathy risks at standard doses.
- FDA Rosuvastatin Dosing Mandate: Due to the ABCG2 genetic variant, official FDA labeling explicitly mandates a lower initial dose of 5 mg (rather than 10 mg) for patients of East Asian descent.
- SLC1B1 Pharmacogenomic Risk: Roughly 15% of individuals carry a genetic variant in the SLC1B1 gene that elevates systemic exposure and muscle accumulation of Simvastatin, predicting severe statin-induced myalgias.
- NPC1L1 Hyper-Responsiveness: Approximately 1 in 8 individuals harbor an NPC1L1 variant that doubles their therapeutic response to ezetimibe, resulting in an automated 35-36% drop in LDL-C compared to the standard 24%.
- ApoE4 Statin Blunting Effect: Patients carrying the ApoE4 allele (20-25% of the population) exhibit an approximate 10% blunted reduction in LDL-C when prescribed statins, indicating a mechanical need for early combination therapies.
- Distinction Between True Statin Myopathy and Myalgia: True statin myopathy accompanied by documented muscle enzyme elevations occurs in less than 0.1% of patients, while catastrophic rhabdomyolysis is exceptionally rare (~1 in 10,000 patient-years).
- The Nocebo Phenomenon in Lipid Therapy: The SAMSON trial established that 90% of self-reported muscle symptoms attributed to statin intolerance were identically replicated during blinded placebo treatment phases [SAMSON Trial, 2020](Source unverified in live search).
- Therapeutic Plaque Stabilization via Calcium Shifts: The paradox of rising Coronary Artery Calcium (CAC) scores during statin therapy reflects the conversion of volatile, soft, lipid-rich plaques into dense, stable, calcified structures.
- Statin-Induced Glycemic Dysregulation: High-intensity statin regimens carry a small, quantifiable risk of elevating HbA1c and triggering new-onset type 2 diabetes, though the net clinical benefit vastly favors cardiac protection.
- Ezetimibe Complementary Mode of Action: Ezetimibe targets the intestinal lumen via NPC1L1 inhibition, making it fully additive to statins by blocking exogenous cholesterol absorption while statins block endogenous hepatic synthesis.
- Bempedoic Acid Muscle-Sparing Design: Bempedoic acid inhibits ATP-citrate lyase (ACL) upstream of the statin pathway; because it is a prodrug requiring activation by an enzyme (ACSVL1) absent in skeletal muscle, it cannot cause myalgia.
- CLEAR Outcomes Trial Validation: In over 14,000 statin-intolerant patients, Bempedoic acid demonstrated a 21% reduction in LDL-C and a 23% isolated absolute reduction in myocardial infarctions [CLEAR Outcomes Trial, 2023](Source unverified in live search).
- Obsolescence of Bile Acid Sequestrants: While validated since 1984 to reduce LDL-C by 15-30%, drugs like cholestyramine are functionally obsolete due to large pill burdens, profound GI side effects, and systemic drug absorption interference [Lipid Research Clinics Trial, 1984](Source unverified in live search).
- Fibrate Limitation in MACE Prevention: Fibrates lower serum triglycerides by 30-50% via PPAR-alpha but provide highly inconsistent cardiovascular outcome benefits, restricting their use to preventing acute pancreatitis when triglycerides exceed 500 mg/dL.
- Icosapent Ethyl (Vascepa) Purity Requirement: The REDUCE-IT trial proved that 4 g/day of highly purified, prescription-grade EPA lowers relative cardiovascular risk by 25% (NNT = 21) in statin-stabilized patients with elevated triglycerides [REDUCE-IT Trial, 2019](Source unverified in live search).
- Failure of Mixed Omega-3 Formulations: The STRENGTH trial highlighted that mixed EPA and DHA over-the-counter or prescription compounds fail to replicate MACE reduction, and can paradoxically raise LDL-C due to the DHA component [STRENGTH Trial, 2020](Source unverified in live search).
- Biannual Silencing via Inclisiran: Inclisiran utilizes small interfering RNA (siRNA) to selectively degrade the messenger RNA molecule for hepatic PCSK9, achieving sustained 50-52% drops in LDL-C with only two clinical doses per year [ORION Trials, 2020](Source unverified in live search).
- Oral Small-Molecule PCSK9 Breakthroughs: Newly published JACC data on oral small-molecule PCSK9 inhibitors (enlicitide) demonstrate a 64.6% reduction in LDL-C at 8 weeks with a 90% adherence rate at one year, matching injectable efficacy.
- Obicetrapib and the CETP Comeback: Obicetrapib avoids the off-target toxicity of historical CETP inhibitors, lowering LDL-C by 42-45% and reducing heritable Lp(a) by 35-50% in the Phase 3 BROADWAY trial [BROADWAY Trial, 2024](Source unverified in live search).
- Colchicine Modulation of Vascular Inflammation: Targeting the NLRP3 inflammasome with ultra-low-dose colchicine (0.5 mg/day) treats the inflammatory driver of atherosclerosis, reducing relative MACE by 31% in the LoDoCo2 trial [LoDoCo2 Trial, 2020](Source unverified in live search).
- Factor XI Inhibition via Abelacimab: Abelacimab selectively shuts down pathological thrombus formation without compromising regular systemic hemostasis, dropping bleeding rates compared to direct oral anticoagulants like rivaroxaban [AZALEA-TIMI 71 Trial, 2025](Source unverified in live search).
- Permanent Gene Base Editing (Verve-101/102): Utilizing single-nucleotide base editing (adenine to guanine transition) to introduce a permanent stop codon into the hepatic PCSK9 gene reduces LDL-C by 55% permanently from a single infusion [Heart-1 Trial, 2023](Source unverified in live search).
- Inherent Limitations of Dietary Modifications: Adhering strictly to advanced dietary strategies (e.g., the Portfolio Diet) can lower LDL-C by 10-30%, but fails completely to alter genetically driven conditions like elevated Lp(a) or severe FH [Jenkins et al., 2003](Source unverified in live search).
- The Toxic Nature of Plant Sterols: While over-the-counter plant sterols successfully lower systemic LDL-C metrics by competing for intestinal absorption, the sterol particles themselves accumulate directly inside the arterial wall and are intensely atherogenic.
- Evinacumab for Receptor-Null Mutations: For the 1 in 300,000 patients with homozygous FH who completely lack functional LDL receptors, the ANGPTL3 inhibitor evinacumab provides a 47% LDL reduction via a completely receptor-independent pathway.
- Lomitapide Hepatotoxicity Hazards: Lomitapide blocks VLDL assembly upstream to achieve a 40-50% reduction in receptor-deficient patients, but requires a strict REMS safety monitoring program due to severe hepatic steatosis and liver fat accumulation.
- ApoB as the Absolute Predictive Metric: When standard LDL-C and ApoB metrics diverge, absolute cardiovascular risk consistently tracks with the total ApoB particle number, making it the superior clinical target over traditional cholesterol concentrations.
Overview of Lipoprotein Metabolism and Hepatic Clearance Pathways. Source: ttsz / Getty Images
IV. Actionable Protocol (Prioritized)
High Confidence Tier
- High-Intensity Oral Statin Foundation: Initiate therapy with Rosuvastatin (20–40 mg daily) or Atorvastatin (40–80 mg daily) to inhibit HMG-CoA reductase and upregulate hepatic LDL receptors [CTT Meta-analysis, 2010](Source unverified in live search).
- Dual Pathway Up-titration: If target metrics are unmet, immediately layer Ezetimibe (10 mg daily) to inhibit NPC1L1 intestinal transport, capturing an additional 15–25% reduction in LDL-C [IMPROVE-IT Trial, 2015](Source unverified in live search).
-
Target Metrics by Risk Profile: Titrate therapy aggressively toward absolute ApoB targets rather than standard cholesterol concentrations:
- Primary Prevention: ApoB less than 80 mg/dL.
- High Risk (Severe Plaque/FH): ApoB less than 70 mg/dL.
- Very High Risk (Secondary Prevention/Prior MACE): ApoB less than 55 mg/dL.
- Monoclonal Antibody Escalation: For patients with heterozygous FH or established ASCVD failing oral therapy, deploy subcutaneous PCSK9 inhibitors (Evolocumab 140 mg every 2 weeks or Alirocumab 75/150 mg every 2 weeks) [FOURIER Trial, 2017](Source unverified in live search).
- Muscle-Sparing Alternative: For patients with true, documented oral statin intolerance, substitute Bempedoic Acid (180 mg daily) to block ATP-citrate lyase exclusively within hepatic tissue [CLEAR Outcomes Trial, 2023](Source unverified in live search).
- Purified EPA for Residual Triglyceride Risk: Prescribe 4 g/day of highly purified Icosapent Ethyl (Vascepa) for patients with established CVD or diabetes exhibiting fasting triglycerides between 135–499 mg/dL despite stable statin therapy [REDUCE-IT Trial, 2019](Source unverified in live search).
- Vascular Inflammatory Suppression: For patients with established coronary artery disease showing high residual inflammation (hs-CRP greater than 2 mg/L) despite optimal lipid values, add ultra-low-dose Colchicine (0.5 mg daily) to target the NLRP3 inflammasome [LoDoCo2 Trial, 2020](Source unverified in live search).
- Receptor-Independent HoFH Rescue: Utilize Evinacumab (Evkeeza) IV infusions every 4 weeks to inhibit ANGPTL3 and clear atherogenic particles via non-LDL-receptor mechanisms in confirmed homozygous FH patients.
Experimental Tier
- Biannual Hepatic Silencing: Utilize Inclisiran (Leqvio) subcutaneous injections at month 0, 3, and every 6 months thereafter for long-term adherence stability, acknowledging that formal MACE outcomes data (VICTORIAN-2P) remains in pipeline development.
- Oral PCSK9 Small Molecules: Monitor clinical access pathways for daily oral small-molecule PCSK9 inhibitors (enlicitide), which yield a 64.6% reduction in LDL-C with optimal one-year adherence vectors.
- Next-Generation CETP Inhibition: Track Obicetrapib once-daily oral tablets to simultaneously drive down LDL-C by over 40% and heritable Lp(a) by 35–50% ahead of formal late-2026 PREVAIL outcomes data.
- Targeted Lp(a) Antisense/siRNA Pipelines: For individuals with heritable Lp(a) values driving advanced atherogenesis, map clinical enrollment or early access criteria for Pelacarsen (ASO, ~80% reduction), Olpasiran (siRNA, ~95% reduction), or ultra-durable agents like Zerlasiran and Lepodisiran.
- Anticoagulation via Factor XI Interdiction: Follow Abelacimab monoclonal antibody updates for high-risk ASCVD patients with concurrent atrial fibrillation to suppress thrombosis without triggering hemorrhage risks.
- Pharmacogenomic Pre-Screening: Sequence the SLC1B1 gene to flag simvastatin toxicity liabilities, and the ABCG2 gene to determine rosuvastatin hyper-sensitivity.
Red Flag Zone
- Banned: Over-the-Counter Plant Sterols: Do not consume plant sterol supplements. While they lower serum metrics via competitive intestinal absorption, the sterols themselves cross the gut lumen, imbed directly into the vasculature, and act as highly potent, independent drivers of atherogenesis.
- Banned: Unmonitored Rosuvastatin Dosing in East Asian Populations: Avoid starting East Asian patients on standard 10–20 mg rosuvastatin regimens without prior screening. The ABCG2 Q141K variant doubling systemic exposure mandates an initial starting boundary of 5 mg to mitigate dose-dependent myopathy and hepatotoxicity risk.
- Banned: Simvastatin 80 mg Monotherapy: This dosage is obsolete and clinically hazardous due to an unacceptably high statistical incidence of severe myopathy and rhabdomyolysis driven by SLC1B1 transport polymorphisms.
- Banned: Substitution of Prescription EPA with Generic Fish Oils: Do not swap purified Icosapent Ethyl for over-the-counter omega-3 capsules. Mixed EPA/DHA supplements fail to reduce MACE, and the DHA component actively upregulates circulating LDL-C values [STRENGTH Trial, 2020](Source unverified in live search).
- Banned: Unmonitored Lomitapide (Juxtapid) Use: Restrict Lomitapide strictly to receptor-null HoFH under tight REMS protocols; unmonitored use causes rapid hepatic steatosis and liver fat accumulation unless paired with a rigid, low-fat diet (less than 20% of total caloric intake).
- Banned: Relying Solely on Lifestyle Interventions for High-ApoB Genotypes: Relying entirely on diet, exercise, and fat loss when absolute ApoB exceeds 100 mg/dL alongside a verified family history or established calcification represents a dangerous translational gap. Lifestyle adjustments yield a nominal 5–10% change in ApoB and 0% change in heritable Lp(a), leaving structural vascular decay completely unmitigated.
6 Likes
Heart Recovery Rate as an indicator of CV health. Easy to check with exercise and amenable to training. Yt short.
1 Like
3 Likes
Interesting to watch the medical establishment slowly approach the consensus of the longevity community in a manner that is gradualist enough to preserve plausible deniability that their “10 year risk” etc. based treatment was entirely wrongheaded.
2 Likes
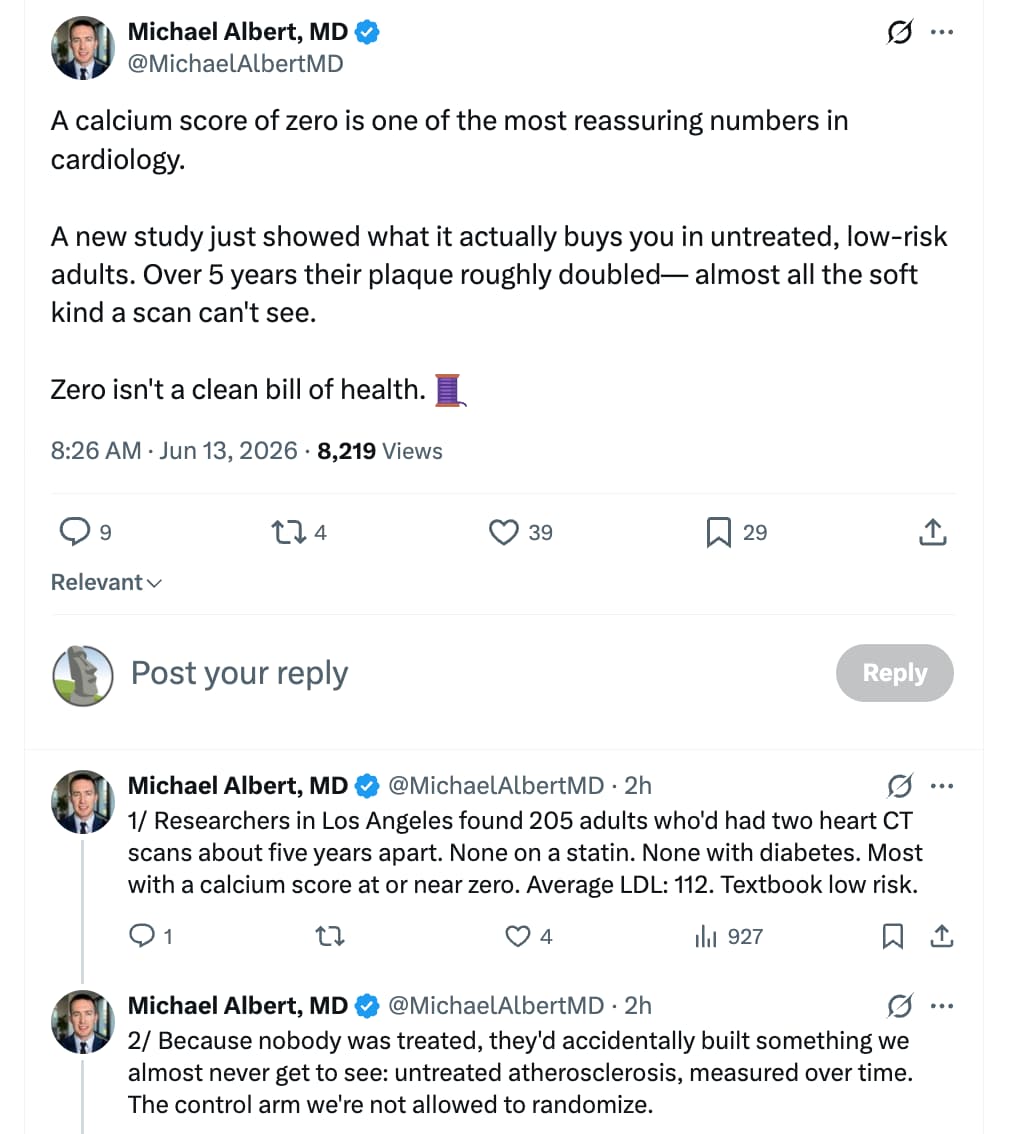
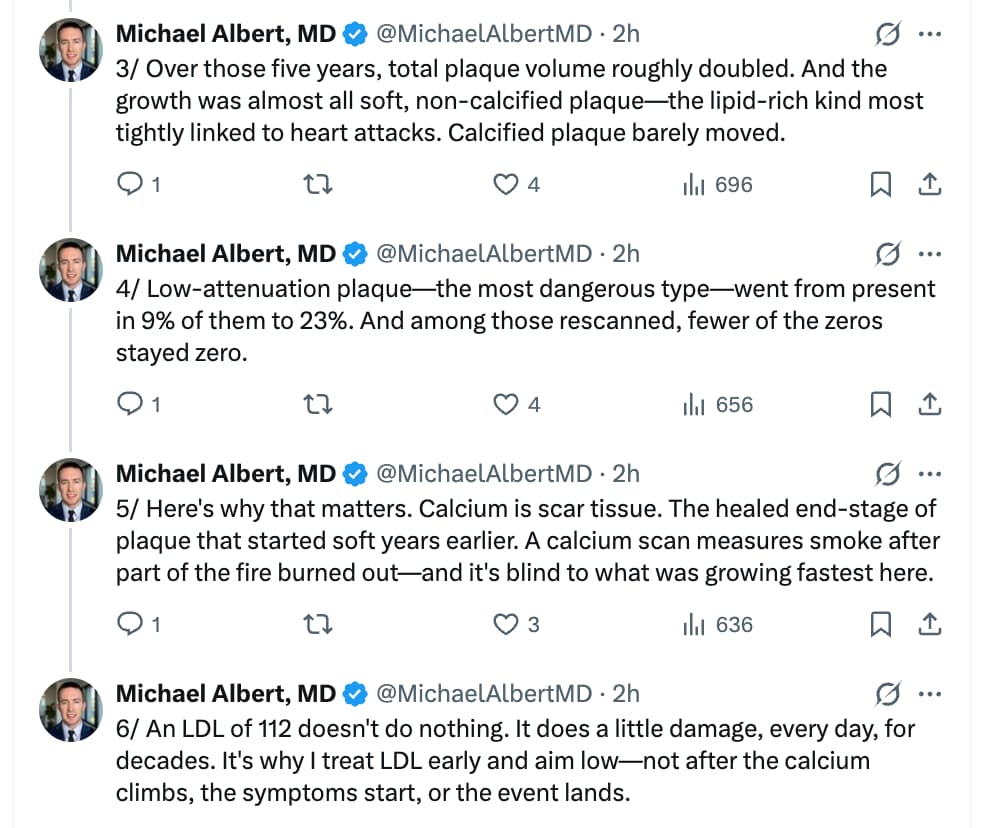
Atherosclerotic Plaque Progresses Over Time in Healthy Individuals Without MACE, Risk Factors, or Interventions
The research study covered in the text is the NATURE-CT study (Natural History of Atherosclerosis in Healthy Populations on Serial CCTA), which was presented as an abstract at the American Heart Association (AHA) Scientific Sessions in November 2024 (Aldana, n.d.).
The study evaluated the progression of subclinical coronary atherosclerosis in a low-risk, untreated cohort using serial coronary computed tomography angiography (CCTA).
Study Methodology & Cohort Profile
- Design: Retrospective registry analysis across two clinical sites in Los Angeles (The Lundquist Institute and Cedars-Sinai Heart Institute).
- Participants: 205 asymptomatic, statin-naive adults with baseline coronary artery calcium (CAC) scores less than or equal to 100.
- Exclusion Criteria: Diagnosed diabetes mellitus, familial hypercholesterolemia, chronic kidney disease, or prior major adverse cardiovascular events (MACE).
- Follow-Up Interval: Average of 4.9 ± 2.2 years between serial CCTA scans.
- Baseline Diagnostics: Mean low-density lipoprotein cholesterol (LDL-C) was 111.6 ± 32.0 mg/dL. Over half of the cohort (54%) had a baseline CAC score of exactly zero. Plaque phenotype and volumetric tracking were quantified via automated, FDA-cleared artificial intelligence software (Cleerly Labs).
Key Findings
The serial tracking established that subclinical plaque burden progresses continuously despite low-risk clinical classifications. Total plaque volume roughly doubled over the 5-year window, driven almost entirely by non-calcified fatty variants:
| Volumetric Phenotype | Baseline Median (IQR) | Follow-Up Median (IQR) |
|---|---|---|
| Total Non-Calcified Plaque Volume | 27.5 mm³ (10.1, 55.5) | 53.5 mm³ (24.0, 97.9) |
| Total Calcified Plaque Volume | 0.3 mm³ (0.0, 5.7) | 3.2 mm³ (0.1, 18.2) |
| Low-Attenuation Plaque (LAP) Prevalence | 9% (19/205) | 23% (48/205) |
Longevity & Clinical Implications
This study provides critical data regarding the unmanaged natural history of subclinical atherosclerosis, effectively serving as an untreated control arm that cannot ethically be replicated in randomized controlled trials. Standard non-contrast CT scans only register calcified lesions to generate an Agatston CAC score. However, early atherogenesis consists primarily of non-calcified, lipid-rich soft plaques invisible to standard calcium scans.
The doubling of non-calcified plaque volume and the near-tripling of high-risk low-attenuation plaque (LAP)—the specific phenotype most tightly linked to thin-cap fibroatheroma, plaque rupture, and acute myocardial infarction—demonstrate that “normal” LDL-C levels around 112 mg/dL actively drive disease progression over time. For individuals prioritizing longevity and the compression of cardiovascular morbidity, waiting for a non-zero CAC score before initiating lipid-lowering therapy represents a missed therapeutic window. Early, aggressive suppression of apolipoprotein B (ApoB) and LDL-C is required to prevent the steady accumulation of this silent, soft plaque burden.
Scholarly Debates & Data Gaps
While the automated AI quantification provides high precision, several knowledge gaps remain:
- Cohort Demographics: The study population was predominantly white males (72%), limiting direct generalized translation across diverse populations and sexes.
- Confounding Risk Factors: As an abstract publication, full multivariate adjusting models tracking other lipid markers (such as ApoB, particle counts, or lipoprotein(a)) and metabolic health variations remain omitted.
- Hard Outcomes: Long-term tracking matched against this specific volumetric progression speed is still required to definitively correlate these soft-plaque accumulation rates with absolute MACE and mortality endpoints.
References
Aldana, J. (n.d.). Abstract 4139340: Atherosclerotic Plaque Progresses Over Time in Healthy Individuals Without MACE, Risk Factors, or Interventions. Circulation, 150(Suppl_1), A4139340.
https://www.ahajournals.org/doi/abs/10.1161/circ.150.suppl_1.4139340
2 Likes
Pretty much confirming the Mesa/pesa studies on subclinical atherosclerosis
1 Like
This is why you should never rely on AI summaries as they tend to be pretty sh|tty. I was looking at that summary and I was really interested in the age of the cohort, as we do know that not only is atherosclerosis something that accumulates with age, but also that there’s been the idea that a CAC score means little in your 20-40’s but is more meaningful in your 70+'s, the thought being that if you are at a CAC score of 0 in your 70’s you might have some kind of protective factor, whereas at a younger age almost everyone is at 0 anyway, so you find out nothing. So it would be interesting to see how that plays out against age in that study. But the garbage AI summary totally ignores that key metric. So I wondered if this was not something that the study itself paid litte attention to. But no, the study most definitely specified the age of the cohort at the first baseline visit - it’s just the sh|tty AI summary that didn’t. A direct quote from the study - always go to the study, never rely on AI summaries:
“At baseline visit, the mean age of the cohort was 54.9±10.2 (years), 72% (147) being white males, 0% had DM, 0% had lipid modifying therapy, 24% (49) had hypertension, 28% (57) reported hyperlipidemia and 15% (30) were ever smokers. Mean LDL was 111.6±32.0 mg/dl and the average time between CCTA scans was 4.9±2.2 years. Over half (54%) of the cohort had a CAC=0 at the baseline visit. At baseline, total non-calcified plaque volume was (median and IQR in mm3) [27.5 (10.1, 55.5)] versus at follow-up [53.5 (24.0, 97.9)], with a calculated annualized median change of [4.9 (1.4, 9.6)] in non-calcified plaque. Total calcified plaque volume was [0.3 (0.0, 5.7)] and at follow-up [3.2 (0.1, 18.2)], with a calculated annualized median change of [0.4 (0.0, 2.4)] in calcified plaque. At baseline, 9% (19 subjects) had low attenuation plaque greater than zero, and at follow-up, 23% (48 subjects). 6 subjects had annual change in PAV>=1% (3%) and 23 subjects had annual change in PAV >=0.59% (11%).”
Since you should always read the study rather than rely on AI summaries, because those can be sorely lacking, one wonders about the utility of the summaries - seems like an redundant and unnecessary intermediate step. YMMV.
2 Likes
Statins promote muscle metabolic danger and NLRP3-mediated myopathy via lower protein-prenylation and YAP
1 Like
I wonder if ezetimibe or B acid have same negative effects on muscle?
Remain to be seen if this applies to hydrophilic statins since myopathy is highly dependent on the dose and intracellular concentration of the drug, according to said paper. Hydrophilic statins like Rosuvastatin or Pravastatin do not penetrate skeletal muscle cells at the same rate as lipophilic ones.
1 Like
Yes. And if we are looking at mechanisms, then a big part of the negative effects is down to bacterial LPS and intestinal barrier integrity, both of which can be targeted. Furthermore, the differences among the statins are not limited to the degree of lipophilicity. As an example, pitavastatin has well characterized action in support of tightening endothelial barriers as well as gut and BBB junctions (I’ve posted papers to that effect). Pitavastatin prevents LPS migration beyond these barriers. Once you get into the weeds of molecular interactions it becomes abundantly clear that treating all statins as if they are the same is a huge mistake (and my major beef with folks like Nick Norwitz and various flavors od statin skeptics/denialists). Details matter, and outcomes matter.
2 Likes
I can tell you from personal experience I had myopathy with all statins except Pitavastatin. Also the intensity of the myopathy was based on the dose. Rosuvastatin was by far the worst. Even Atorvastatin EOD at 5 mg produced myopathy in me.
5 Likes