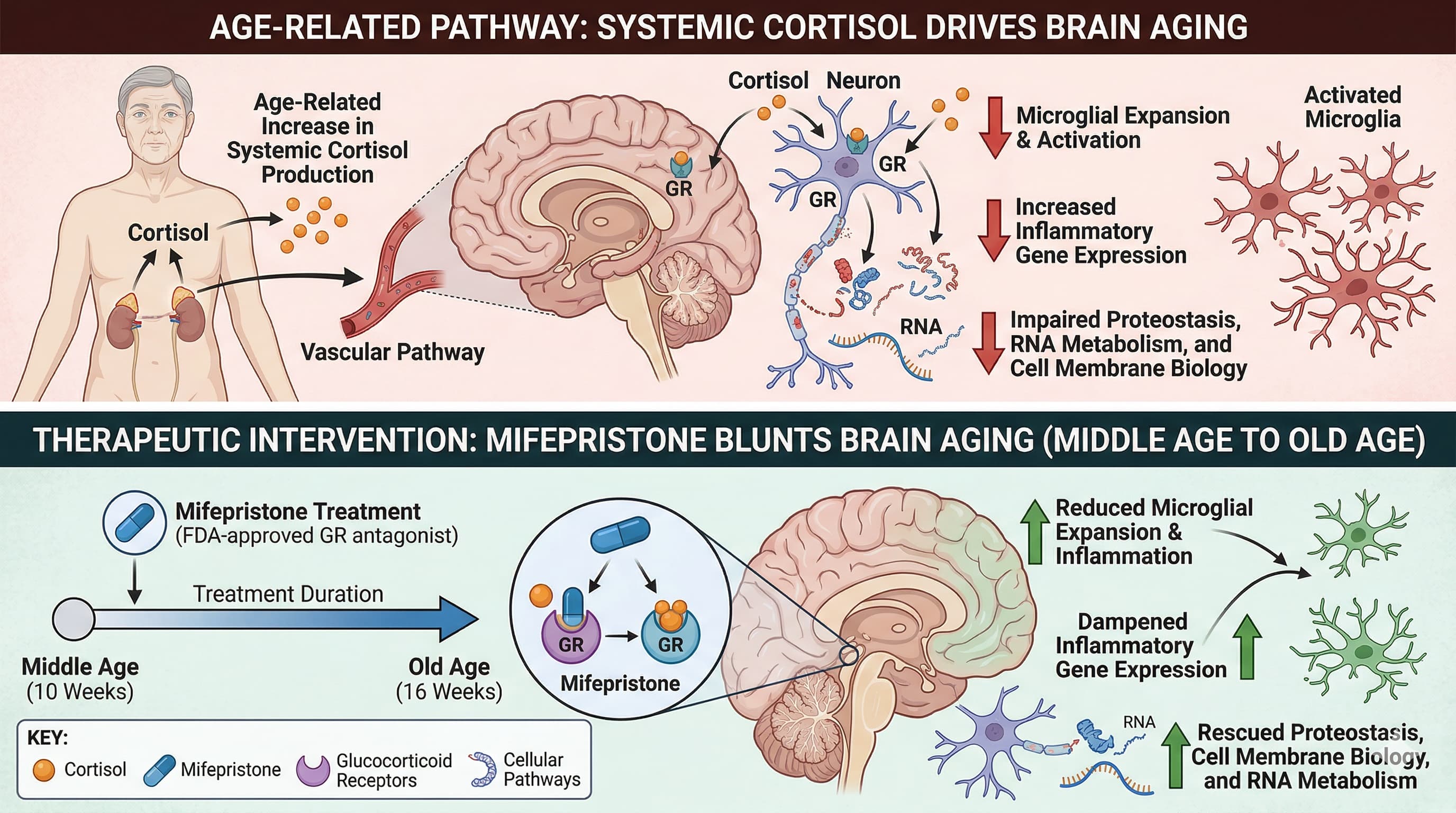
Follow-on Gemini 3 Pro Prompt:
Is there any evidence that pulsatile dosing of mifepristone may be effective in minimizing brain inflammation, while also moderating disruption of the HPA axis, significant hypokalemia risk, and catastrophic CYP3A4 suppression.
Gemini 3 Pro Response:
The short answer is half-yes, half-no. While intermittent or “short-course” dosing elegantly solves the endocrine issues and still appears to reset brain inflammation, it completely fails to solve the CYP3A4 interaction risk. Here is how the data breaks down when we separate the endocrine pharmacology from the hepatic pharmacokinetics.
The Successes: Inflammation and the HPA Axis
There is actually excellent evidence that the brain does not need chronic glucocorticoid blockade to benefit; it just needs a circuit breaker.
-
Minimizing Brain Inflammation: Pre-clinical models of Gulf War Illness (a condition characterized by severe, stress-induced microglial activation) have successfully used intermittent or short-term post-exposure dosing of mifepristone (often combined with the anti-inflammatory etanercept). The data suggests that short bursts of GR antagonism are enough to “reset” the aberrant homeostatic signaling of microglia, returning them to a resting state without needing perpetual blockade.
-
Moderating HPA Axis Disruption & Hypokalemia: Human pilot studies on combat-related PTSD support the “pulse” theory. When veterans were given a short course of mifepristone, their cortisol and ACTH predictably spiked (due to blocked negative feedback). However, at a four-week follow-up after the drug was stopped, their HPA axis had successfully recalibrated to a healthier baseline. Pulsing the drug allows the endocrine system to recover between doses, virtually eliminating the risk of the chronic mineralocorticoid spillover that causes hypokalemia.
The Fatal Flaw: Catastrophic CYP3A4 Suppression
This is where the pulsatile strategy falls apart for anyone on a longevity stack.
Mifepristone is not a standard, competitive enzyme inhibitor that simply blocks the door and leaves. It is a mechanism-based “suicide” inhibitor of CYP3A4.
When mifepristone is metabolized by CYP3A4, it creates a reactive intermediate that permanently binds to and destroys the enzyme. Even after the mifepristone has cleared your bloodstream, that specific CYP3A4 enzyme is dead.
To restore your drug-clearing capacity, your liver has to manufacture entirely new CYP3A4 proteins from scratch. The half-life of CYP3A4 enzyme turnover is roughly 36 to 72 hours. Because mifepristone itself has a long serum half-life (~30 hours) due to its high affinity for alpha-1-acid glycoprotein (AAG), the timeline looks like this:
- You take a pulse dose.
- The drug stays in your system for days, destroying CYP3A4.
- Once the drug is gone, it takes another 3 to 6 days for the liver to rebuild its enzyme inventory.
The Bottom Line: If you pulse mifepristone even once a week, you are keeping your liver in a near-continuous state of CYP3A4 paralysis. You would still face severe, potentially life-threatening toxicity risks if you co-administered it with CYP3A4 substrates like Rapamycin, Atorvastatin, or PDE5 inhibitors.
The Verdict: Pulsatile dosing of mifepristone is an elegant neuroendocrine strategy, but a pharmacokinetic trap. Until we have highly targeted delivery methods (like intrathecal administration or non-systemic prodrugs), it cannot be safely integrated into a multi-drug longevity protocol.
If you want to pull microglia out of their neurotoxic, highly activated states (DAM/IRM) without paralyzing your liver’s CYP3A4 pathways, you have to abandon most of the popular longevity botanicals. Fisetin, Quercetin, Curcumin, and Apigenin are all notorious, promiscuous CYP inhibitors.
To safely stack neuro-anti-inflammatories alongside rapamycin or statins, you need compounds that bypass the CYP450 system entirely. Here are the four most validated, CYP3A4-safe targets for dampening microglial activation.
1. Low-Dose Naltrexone (LDN)
The direct pharmacological off-switch for microglia.
At standard addiction-treatment doses (50mg), naltrexone is an opioid antagonist. But at very low doses (1.5 to 4.5mg), it exhibits a completely different pharmacological profile: it acts as an antagonist at Toll-like Receptor 4 (TLR4).
TLR4 is the primary “danger sensing” receptor on the surface of microglia. When activated by cellular debris or systemic inflammation, TLR4 locks microglia into the M1 (pro-inflammatory) state. LDN physically blocks TLR4, halting the inflammatory cascade and allowing microglia to return to their M2 (homeostatic/repair) phenotype.
-
Metabolism: Naltrexone is primarily metabolized by non-cytochrome cytosolic enzymes (aldo-keto reductases). It does not induce or inhibit CYP3A4.
-
Validation: Level B/C. Extensively used off-label in clinical neurology for MS, fibromyalgia, and Long-COVID neuroinflammation.
-
The Protocol: Typically titrated slowly. Start at 1.5mg taken at night, increasing by 1.5mg every two weeks up to a maximum of 4.5mg.
2. Exogenous D-Beta-Hydroxybutyrate (BHB)
The epigenetic inflammasome blocker.
Ketones are usually discussed as an alternative brain fuel, but their longevity value is actually signaling. The specific enantiomer D-BHB directly inhibits the NLRP3 inflammasome—the intracellular complex responsible for churning out inflammatory cytokines (like IL-1β) in aging microglia.
Furthermore, D-BHB acts as an endogenous Histone Deacetylase (HDAC) inhibitor, epigenetically upregulating BDNF (Brain-Derived Neurotrophic Factor) while simultaneously suppressing inflammatory gene transcription in glial cells.
-
Metabolism: BHB is a natural metabolite; it clears via cellular respiration and does not interact with hepatic CYPs.
-
Validation: Level B/C. Strong pre-clinical data for neuroprotection; emerging human clinical trials show cognitive rescue in Mild Cognitive Impairment (MCI).
-
The Protocol: Exogenous ketone esters (not salts, which carry a heavy sodium load) dosed to achieve a blood BHB concentration of 1.5 to 2.5 mmol/L during periods of deep work or fasting.
3. Spermidine
The glial autophagy restorer.
One of the primary reasons microglia become chronically activated with age is that their internal “garbage disposal” breaks down. They accumulate lipofuscin, misfolded proteins, and damaged mitochondria. Spermidine, a natural polyamine, restores this via the hypusination of the translation factor eIF5A. This jumpstarts autophagic flux, allowing microglia to digest their internal waste and quiet down.
-
Metabolism: Polyamines are metabolized by specific oxidases (e.g., SMOX) and acetyltransferases, safely bypassing the CYP450 system.
-
Validation: Level B/C. High dietary spermidine correlates strongly with reduced cortical atrophy in humans, and robust lifespan extension/neuroprotection is seen in mouse models.
-
The Protocol: 3mg to 6mg daily, typically sourced from concentrated wheat germ extract. Must be taken consistently for months to alter intracellular polyamine pools.
4. Exertional Lactate (The Physiological Route)
The endogenous neuro-calmer.
For years, lactate was viewed as a toxic byproduct of anaerobic metabolism. It is actually a potent neurohormone. During vigorous exercise (upper Zone 2 and Zone 5 HIIT), peripheral lactate crosses the blood-brain barrier and binds to a specific receptor on microglia called HCAR1 (Hydroxycarboxylic acid receptor 1).
Activation of HCAR1 forcefully suppresses microglial activation and drives the production of VEGF (Vascular Endothelial Growth Factor), promoting new blood vessel growth in the hippocampus.
-
Metabolism: Cleared by the liver and heart (Cori cycle); zero drug-drug interactions.
-
Validation: Level A. This mechanism is one of the primary reasons cardiovascular exercise is the single most validated intervention for preventing neurodegeneration.
-
The Protocol: 30-45 minutes of exercise that pushes blood lactate to roughly 2.0 - 4.0 mmol/L (the lactate turnpoint), achieved via sustained Zone 2 work or high-intensity intervals.
The Botanical Warning: If you are building a strict longevity stack around CYP3A4 substrates like Rapamycin, do not attempt to lower neuroinflammation using high-dose plant polyphenols. Apigenin, Quercetin, Fisetin, and Curcumin are all clinically significant inhibitors of CYP3A4, CYP2C9, or CYP1A2, and will cause unpredictable and potentially dangerous spikes in your pharmaceutical blood levels.