Glucose dysregulation is an extremely complex topic that involves much more than carbohydrate consumption and insulin resistance.
I have had the idea of doing a comprehensive deep dive on the topic since I listened to Dr DeFronzo talks about the “Ominous Octet” (the eight underlying pathologies of glucose dysregulation). Since that original 2009 list, the research has expanded to the “Egregious Eleven” (Schwartz et al., 2016), and we have now identified a total of 21 pathologies.
I initially used ChatGPT’s Deep Research feature when it came out but, due to of the complexity of this task, the results were not convincing at that time. I have since switched to Claude Opus 4.6, which proved much more effective this time.
BTW The project was extensive enough to reach my monthly tokens limit at once.
To keep the information actionable and of a reasonable length, I have structured the results into five separate documents, which I will share in upcoming posts:
Your complete five-document set is now:
- Historical Overview & Master Reference List
- Differential Diagnostic Protocol
- Pharmacological Reference
And for those who want to even dig more into the subject
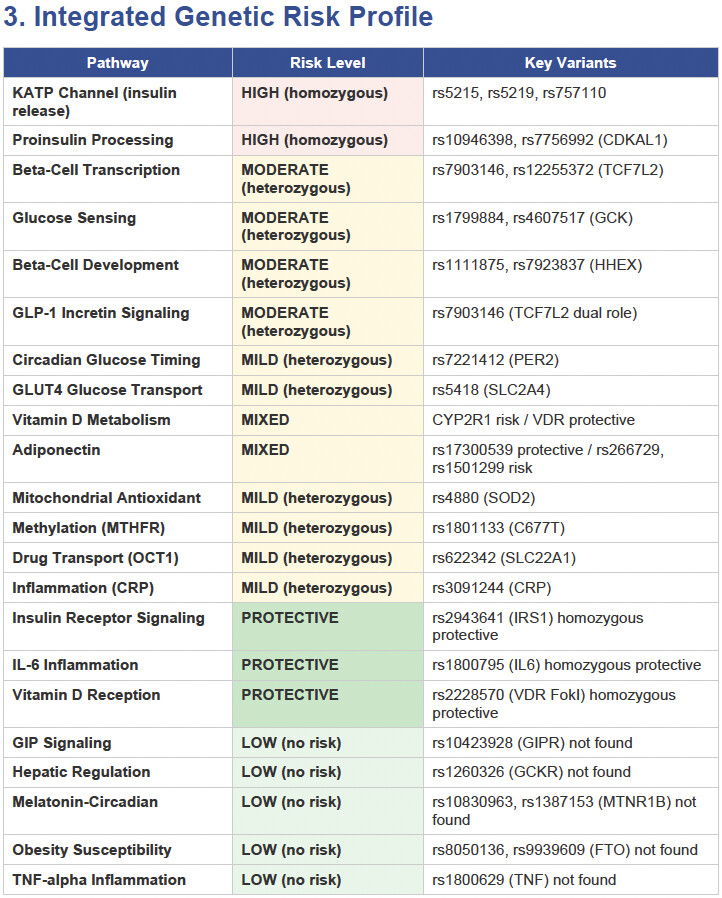
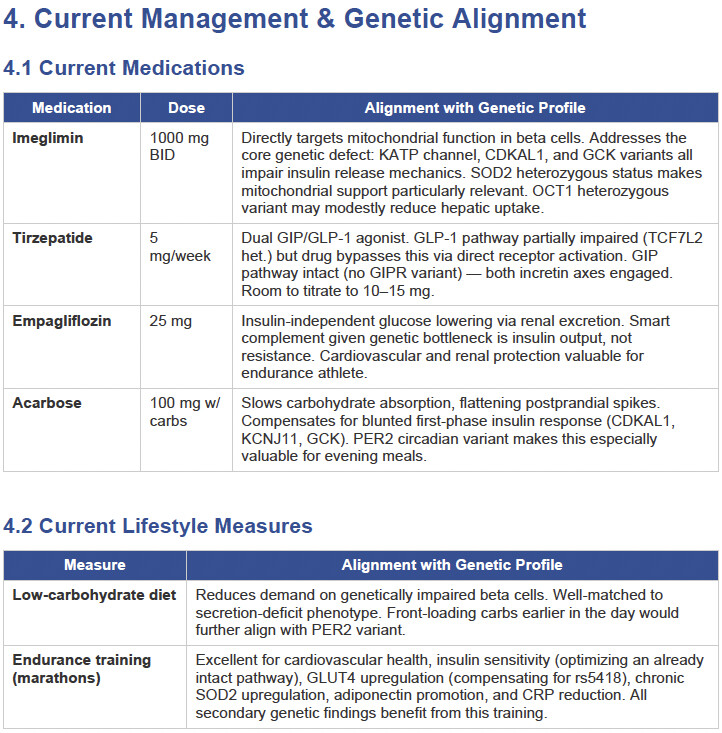
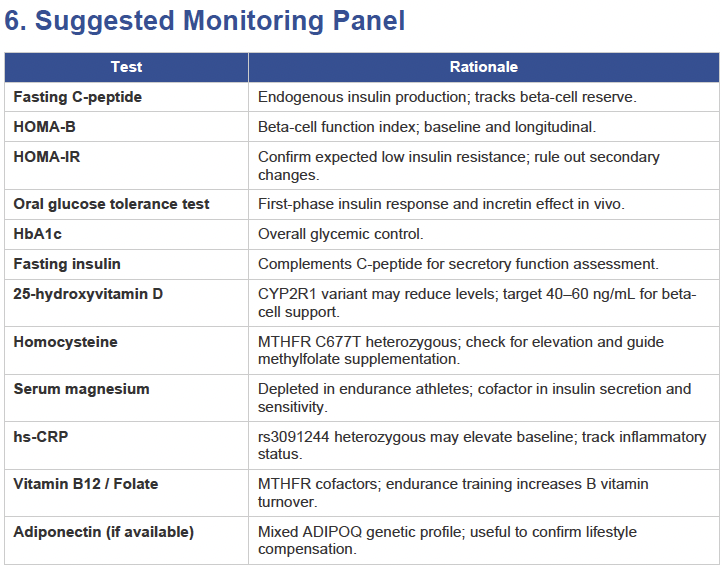
Then I applied all those findings to get extremely actionable insights in my N of 1 case.
To get started, here is the list of the pathologies.
The Original Ominous Octet (DeFronzo, 2009):
- Pancreatic β-cell dysfunction — Progressive failure of insulin secretion; the central defect upon which all others converge.
- Pancreatic α-cell dysfunction — Inappropriate glucagon secretion that drives excess hepatic glucose output, particularly in the fasting state.
- Hepatic glucose overproduction (Liver) — Increased gluconeogenesis and glycogenolysis due to hepatic insulin resistance and excess glucagon signaling.
- Skeletal muscle insulin resistance — Impaired glucose uptake in the body’s largest insulin-responsive tissue.
- Adipose tissue dysfunction / increased lipolysis — Accelerated free fatty acid release causing lipotoxicity in β-cells, liver, and muscle; also a source of inflammatory adipokines.
- Decreased incretin effect (GI tract) — Diminished action of GLP-1 and GIP, reducing meal-stimulated insulin amplification.
- Increased renal glucose reabsorption (Kidney) — Upregulated SGLT2 transporters raise the renal threshold for glucose, perpetuating hyperglycemia.
- Brain / neurotransmitter dysfunction — Hypothalamic insulin resistance, impaired satiety signaling, and central appetite dysregulation promoting overeating.
Additions in the Egregious Eleven (Schwartz et al., 2016):
- Immune dysregulation / chronic inflammation — Low-grade inflammation with elevated TNF-α, IL-6, and other cytokines from visceral fat and immune cells, contributing to both insulin resistance and β-cell apoptosis.
- Altered gut microbiome (Colon) — Dysbiosis affecting bile acid metabolism, short-chain fatty acid production, gut permeability, and systemic inflammation, all influencing glucose homeostasis.
- Stomach / small intestine dysfunction (decreased amylin) — As β-cells fail, amylin co-secretion declines, leading to accelerated gastric emptying, exaggerated postprandial glucose spikes, and loss of glucagon suppression.
Additions from the Dirty Dozen (Kalra et al., 2013):
- Dopamine / catecholamine dysregulation — Sustained hyperadrenergic tone and altered central dopaminergic signaling worsen insulin resistance and glycemic control. Bromocriptine QR targets this pathway therapeutically.
- Vitamin D deficiency — Vitamin D modulates both insulin secretion and insulin sensitivity, acts as an immunomodulatory hormone reducing pro-inflammatory cytokines, and cross-talks with the renin-angiotensin system in β-cells.
- Renin-angiotensin system (RAS) overactivity — Angiotensin II signaling generates reactive oxygen species and interferes with insulin signaling pathways; ACE inhibitors and ARBs are associated with lower incidence of new-onset diabetes.
- Testosterone deficiency (in men) / androgen excess (in women) — Low testosterone in men precedes diabetes onset and worsens insulin resistance; androgen deprivation therapy increases diabetes risk. In women, hyperandrogenism (as in PCOS) is associated with metabolic syndrome and insulin resistance.
Additional Emerging Mechanisms (Treacherous Thirteen onward):
- Iron overload / dysregulated iron metabolism — Elevated body iron stores and ferritin are positively associated with T2D and insulin resistance; increased divalent metal transporter 1 (DMT1) activity damages β-cells directly.
- Gut-derived serotonin dysregulation — Peripheral serotonin synthesized in enterochromaffin cells influences hepatic glucose metabolism, adipose tissue function, and β-cell proliferation; its dysregulation has been linked to worsening glycemia.
- Epigenetic modifications — Heritable changes in gene expression (DNA methylation, histone modification) influenced by maternal hyperglycemia, nutrition, and environment that alter insulin signaling gene activity across generations.
- Ectopic lipid deposition (twin-cycle hypothesis) — Excess fat accumulation specifically in the liver and pancreas, as distinct from general adiposity, representing a potentially reversible cause of β-cell failure. This is the basis for Roy Taylor’s remission work demonstrating that targeted fat loss from these organs can restore function.
- Endoplasmic reticulum (ER) stress and oxidative stress — Chronic metabolic overload triggers ER stress and excessive reactive oxygen species in β-cells and insulin-target tissues, activating inflammatory pathways and accelerating cell death.
- Bile acid signaling dysregulation — Bile acids act as metabolic hormones through the FXR and TGR5 receptors, influencing GLP-1 secretion, hepatic glucose production, and energy expenditure; disrupted bile acid metabolism in T2D contributes to impaired incretin signaling and dyslipidemia.
A few important caveats: Mechanisms 1–11 (the Egregious Eleven) represent the most widely cited and clinically operationalized framework, with targeted therapies mapped to each pathway. Mechanisms 12–15 (the Dirty Dozen additions) have strong epidemiological and clinical support but are less uniformly integrated into treatment algorithms. Mechanisms 16–21 are increasingly well-supported in the research literature but are still being characterized in terms of therapeutic targeting. There is also meaningful overlap among many of these — for example, inflammation, oxidative stress, ER stress, and ectopic lipid deposition are deeply interconnected rather than fully independent pathways.
The overarching takeaway remains consistent with the beta-cell-centric model: nearly all of these mechanisms either damage the β-cell directly, result from β-cell failure, or both — reinforcing the case for early, multi-targeted combination therapy.