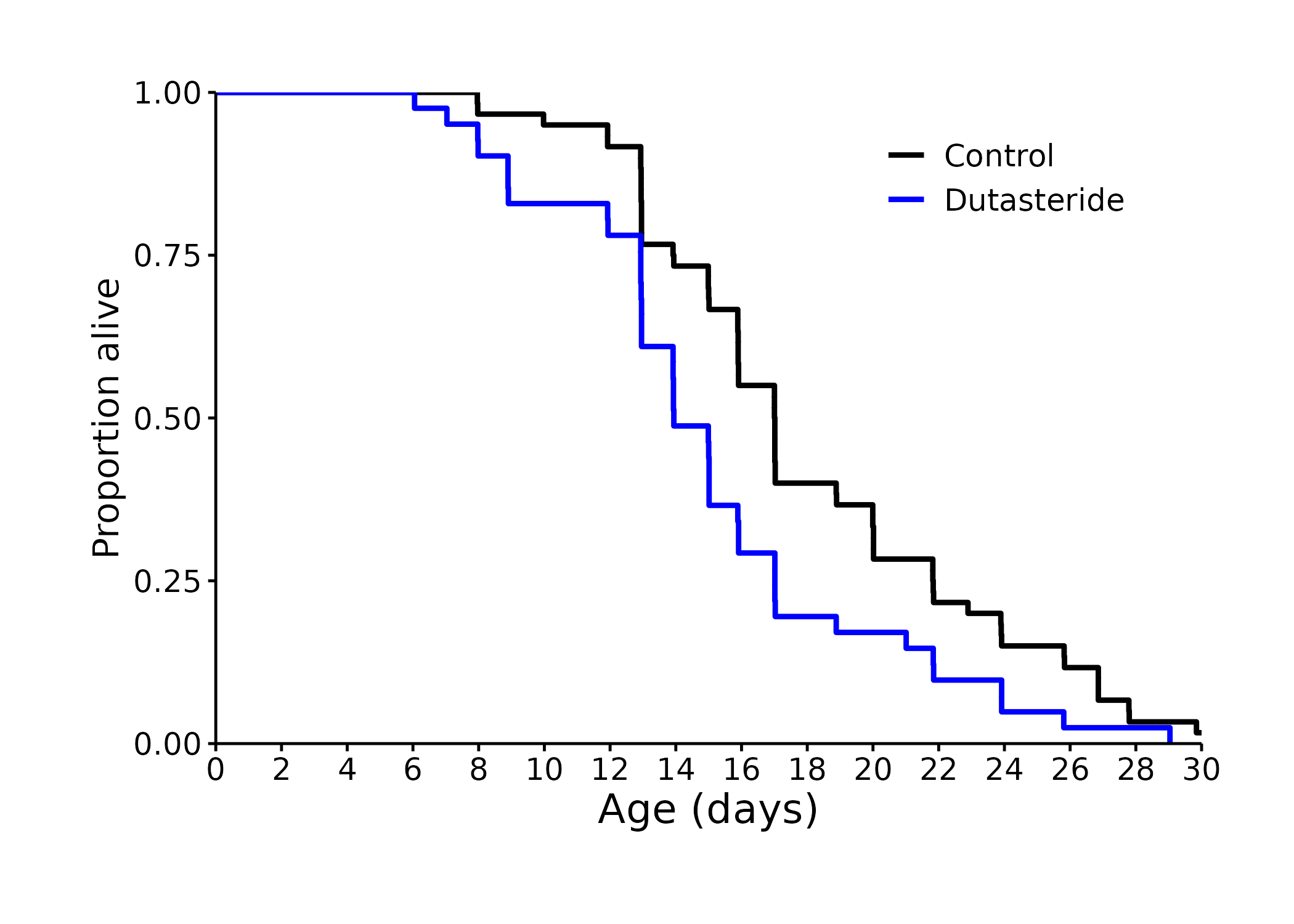
Quoting @RapAdmin 's summary of Peter Attia’s podcast with Richard Miller:
Richard Miller tells Peter Attia that a pharmacologist named James Simpkins has studied estrogen receptors and suggested investigating 17α-Estradiol. He had tried to make a compound that was like estrogen—17β-estradiol—which is generically crudely referred to as estrogen. …
James Simpkins synthesized 17α-Estradiol—very much the same compound except for one of the bonds tilts up instead of tilting down. And in his studies mostly in tissue culture cells, he found that yes, it was tenfold less active compared to 17β-estradiol (the compound which most people refer to as the estrogen used in hormone replacement therapy).
When he gave it to mice, he found it was non-feminizing : So when you give it to male mice, it did not induce the secondary sexual characteristics that the stronger estrogens, like 17 beta- estradiol, estrogen would do. His idea was that giving estrogen to male mice might make them live as long as female mice (female mice live 5-10% longer than males) AND this compound called 17α-Estradiol will be good for males because “no man wants to look feminine” even if he lives as long as a women.
Big caveat: nearly all that follows was done in high fat-fed obese mice, which may not give insight on the mechanisms relevant to its effects in normal-for-one’s-age aging:
the signaling mechanism(s) and primary tissues through which 17α-E2 elicits these benefits remain unknown. Although 17α-E2 is a naturally occurring enantiomer to 17β-estradiol (17β-E2), it has been postulated that 17α-E2 signals through a novel uncharacterized receptor (Toran-Allerand, 2005; Toran-Allerand et al., 2002; Toran-Allerand et al., 2005; Green and Simpkins, 2000) as opposed to classical estrogen receptors α (ERα) and β (ERβ), which is due to 17α-E2 having significantly reduced binding affinity for ERα and ERβ as compared to 17β-E2 (Edwards and McGUIRE, 1980; Korenman, 1969; Littlefield et al., 1990; Anstead et al., 1997). For this reason, 17α-E2 is often referred to as a non-feminizing estrogen (Green and Simpkins, 2000; Engler-Chiurazzi et al., 2017; Kaur et al., 2015). A few studies have suggested that a novel but uncharacterized estrogen receptor, termed ER-X, may mediate 17α-E2 actions in the brain (Toran-Allerand, 2005; Toran-Allerand et al., 2002; Toran-Allerand et al., 2005; Green and Simpkins, 2000), although more recent studies supporting this hypothesis are lacking in the literature. Similarly, no reports to date have directly tested whether the doses of 17α-E2 shown to improve healthspan and lifespan in mice are mediated through ERα and/or ERβ.
There is a multitude of data in the diabetes and metabolism literature demonstrating that ERα is a regulator of systemic metabolic parameters. Although most of these studies have historically been performed in female mammals, more recent studies have demonstrated that ERα also plays a critical role in modulating metabolism in male mammals. For instance, Allard and colleagues recently demonstrated that genomic actions of ERα regulate systemic glucose homeostasis in mice of both sexes and insulin production and release in males (Allard et al., 2019). Other studies have also determined that hepatic steatosis and insulin sensitivity, and therefore the control of gluconeogenesis, are regulated through FOXO1 in an ERα-dependent manner in male mice (Yan et al., 2019). Furthermore, hepatocyte-specific deletion of ERα was sufficient to abrogate similar estrogen-mediated metabolic benefits (Guillaume et al., 2019; Qiu et al., 2017; Meda et al., 2020). Given that several reports have linked the administration of 17α-E2 to improvements in metabolic homeostasis, we hypothesized that 17α-E2 signals through ERα to modulate hepatic function and systemic metabolism, thereby potentially contributing to the lifespan-extending effects of 17α-E2.
Herein, we show that 17α-estradiol elicits similar genomic binding and transcriptional activation through estrogen receptor α (ERα) to that of 17β-estradiol. In addition, we show that the ablation of ERα completely attenuates the beneficial metabolic effects of 17α-E2 in male mice. …
these findings strongly suggest that the liver is a primary site where 17α-E2 acts to improve metabolic homeostasis due to gluconeogenesis being tightly controlled by hormonal actions on hepatocytes (Zhang et al., 2018). However, it also well established that the hypothalamus can directly modulate gluconeogenesis in the liver through hepatic innervation … Interestingly, we found that [acute intracerebroventricular (ICV)] administration of 17α-E2 essentially phenocopied the effects of peripheral 17α-E2 infusion with regard to GIRs and suppression of hepatic gluconeogenesis (Figure 6G–I). These findings suggest that 17α-E2 likely acts through hypothalamic neurons to regulate hepatic gluconeogenesis. Indeed, agouti-related peptide/neuropeptide Y (AgRP/NPY) and pro-opiomelanocortin (Pomc) neurons are known to regulate hepatic glucose production (Könner et al., 2007; Ruud et al., 2017; Pocai et al., 2005a; Pocai et al., 2005b; Dodd et al., 2018) and both neuronal populations express ERα …
Our findings suggest that 17α-E2 may act through the liver and hypothalamus to improve metabolic parameters in male mice. Lastly, we also determined that 17α-E2 improves metabolic parameters in male rats, thereby proving that the beneficial effects of 17α-E2 are not limited to mice. Collectively, these studies suggest ERα may be a drug target for mitigating chronic diseases in male mammals.
https://elifesciences.org/articles/59616
We recently reported that estrogen receptor α is required for the majority of 17α-E2-mediated benefits in male mice, but that 17α-E2 also attenuates fibrogenesis in liver, which is regulated by estrogen receptor β (ERβ)-expressing hepatic stellate cells (HSC). The current studies sought to determine if 17α-E2-mediated benefits on systemic and hepatic metabolism are ERβ-dependent. We found that 17α-E2 treatment reversed obesity and related systemic metabolic sequela in both male and female mice, but this was partially blocked in female, but not male, ERβKO mice. ERβ ablation in male mice attenuated 17α-E2-mediated benefits on hepatic stearoyl-coenyzme A desaturase 1 (SCD1) and transforming growth factor β1 (TGF-β1) production, which play critical roles in HSC activation and liver fibrosis. We also found that 17α-E2 treatment suppresses SCD1 production in cultured hepatocytes and hepatic stellate cells, indicating that 17α-E2 directly signals in both cell-types to suppress drivers of steatosis and fibrosis. We conclude that ERβ partially controls 17α-E2-mediated benefits on systemic metabolic regulation in female, but not male, mice, and that 17α-E2 likely signals through ERβ in HSCs to attenuate pro-fibrotic mechanisms.
https://doi.org/10.1038/s41598-023-37007-1
Work from our lab found that 17α-E2 acts through pro-opiomelanocortin (POMC) appetite- regulating neurons in the hypothalamus, which respond to insulin and induce satiety. However, it is unclear whether 17α-E2 is acting directly on POMC neurons or indirectly through improvements in insulin sensitivity to induce reductions in food intake and adiposity. Discovering this mechanism of action could provide useful potential therapeutic targets for metabolic disorders in elderly obese people.
Purpose: The purpose of this study was to use an insulin-resistant model to determine whether 17α- estradiol (17α-E2) exerts its action via insulin signaling in POMC neurons in the hypothalamus.
Methods: Mice lacking the insulin receptors on POMC neurons (POMC-IR KO) and wild type (WT) mice, were pre-fattened via a 45% high-fat diet (HFD) for about 6 months. Baseline measures were taken, including body mass, daily calorie intake, body composition (lean and fat mass), fasting blood glucose, and fasting insulin, before randomizing the groups. The mice were then treated with either HFD or HFD+17α-E2 (14.4ppm).
Results: When compared to wild type mice with 17α-estradiol, POMC-IR KO mice treated with 17α-E2 demonstrated no significant differences in the percent change in the body mass (-5.91% ± 1.34; NS), fat mass (14.6g ± 1.41; NS), fasting glucose, fasting insulin, glucose tolerance, and insulin sensitivity. There were no significant improvements between Wild type and POMC-IR KO mice treated with high-fat diet.
Discussions/conclusions: The beneficial food intake, adiposity, and metabolic effects of 17α-E2 are not mediated via insulin signaling in POMC neurons. 17α-estradiol may be acting through some other mechanism, such as estrogen receptor α, on POMC neurons or multiple neuronal populations in the brain. However, determining molecular targets of 17α-E2 and their physiological role in curtailing disease conditions needs to be studied in detail.
Full PDF: