https://isitketo.org/category/vegetables
What is the lowest blood glucose level you've ever had? - Ketogenic Forums (keto forums!)
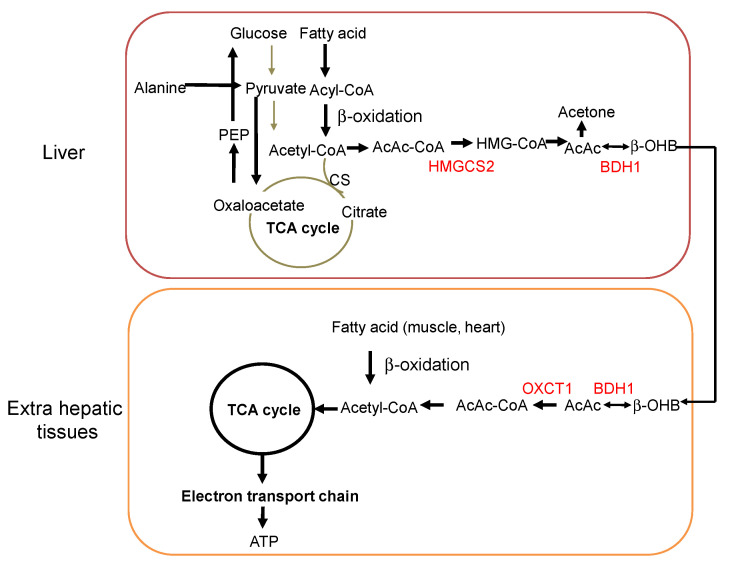
d-beta-hydroxybutyrate vs l-beta-hydroxybutyrate
foods I like: cashew queso [IN GLASS jar], beyond sausage, acai berries, jarred kale, mushrooms, vegan sugar-free chocolate [eg Lily’s], macademia nuts/pecans > almonds > other nuts, avocados, olives, high-polyphenol EVOO (eg Kosterina), tapenade, tofu, bok choy/endives/broccoli rabe/celery/chard >> other veggies, oat fiber, shirataki noodles/konjac, seaweed, nut milks/soymilk,ThinSlim bread, and [yeah] I guess shellfish/crustaceans [though avoid the non-veg* options in favor of veg* when possible]
Watercress/chard/chicories (like endives/escarole) is the best (also very filling and high in nutritional density). archive.ph. The Best Keto Vegetables List - The ultimate Low Carb Vegetables Guide
Rarer: cauliflower hummus
Macedonia [Vitalia] - Home | Vitalia Healthy Food - has special keto granola that’s cheap (https://www.naturalniprodukti.com/en/keto-granola-muesli-with-chocolate-and-coconut). In Europe, there is a wide variety of fake meat/tofu of different brands
It is IMPORTANT to note that nutritional deficiencies can be WAY higher on keto, so get Vitamin supplements, and potassium electrolytes (why aren’t there potassium keto esters yet?)
POSSIBLY SGLT2 inhibitors/metformin are good on the edge (as is anything that could lower blood glucose levels on sum [which may be enough to KEEP you in ketosis in “edge” cases in enough instances to matter, and when doing keto there are A LOT of edge cases])
===
On BHB esters: (The Definitive Guide to Ketone Esters and Ketone Salts - Perfect Keto | Top 7 Exogenous Ketone Supplements (Based on Science))
Ketone salts (usually in the form of D/L-BHB salts). Non-ethanol alcohols (usually in the form of (R)-1,3-butanediol). Ketone esters (usually in the form of a bond between D-BHB and (R)-1,3-butanediol). A combination of D-beta-hydroxybutyric acid and (R)-1,3-butanediol.The difference between the first three has to do with how the ketone bodies are chemically bound to alcohol or salts.
Salts are much easier to manufacture, which is why most exogenous ketone supplements on the market are sold as salts. The problem with salts is that they can lead to stomach cramps and GI irritation, which is why I try to avoid them.
Plus, it’s worth noting that the only bioavailable form of BHB is D-BHB. As a result, products that contain a mix of both L-BHB and D-BHB are usually less effective (depending on the dosage) than products that only contain D-BHB.
Eric Verdin says the salts are worthless [they only work for 1 hour]. Eric uses Science Behind Metabolic Switch Ketone Ester – Juvenescence . His product is this - Metabolic Switch® Powder – Juvenescence. He mentions the book ketotarian (https://www.amazon.com/Ketotarian-Mostly-Plant-Based-Cravings-Inflammation/dp/0525537171/ref=sr_1_1?keywords=ketotarian&qid=1673241702&sr=8-1 )
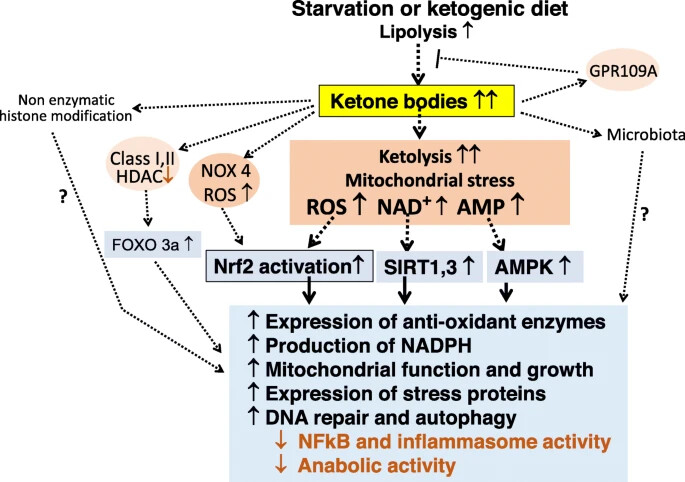
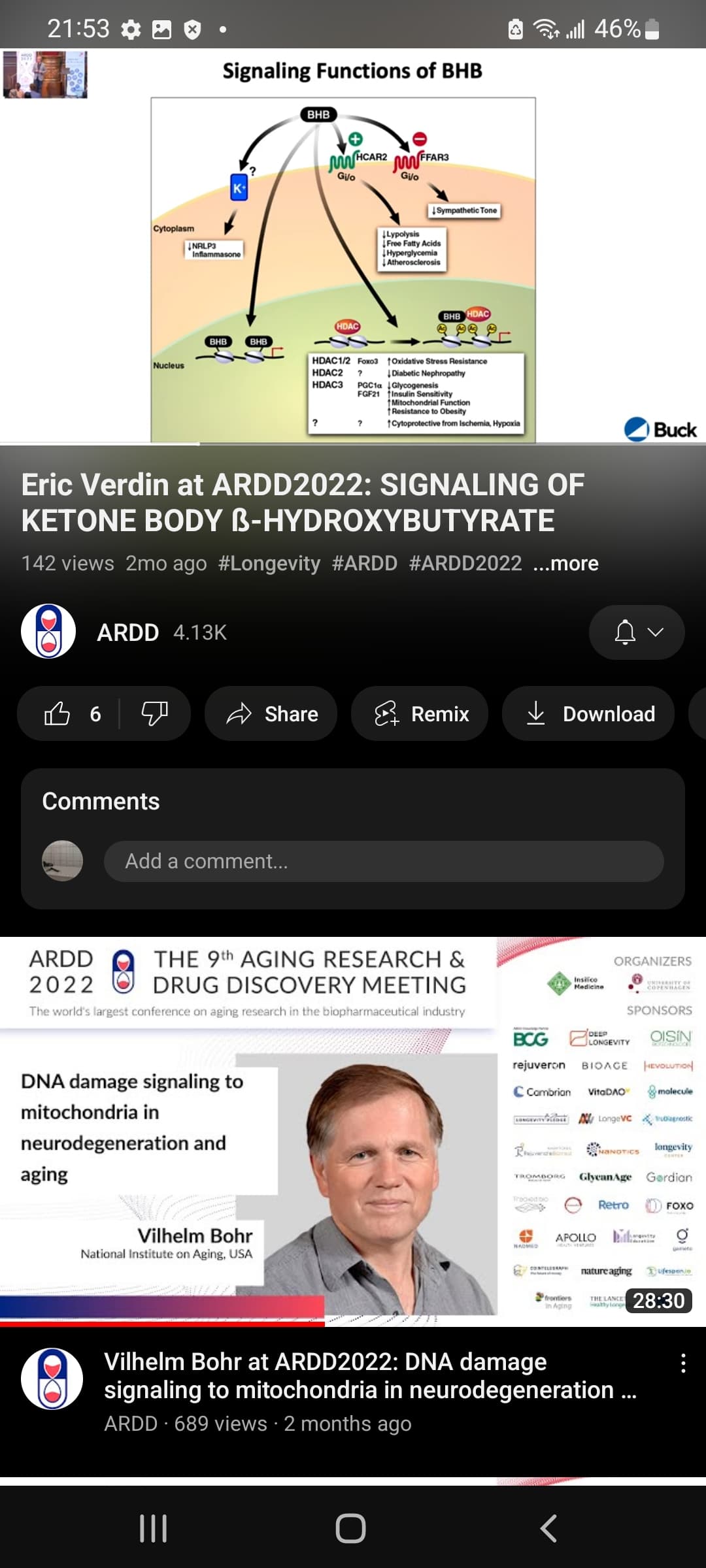
getting into ketosis does not mean keto-adapted (it takes longer for cells to make the transcriptomic adjustments to be properly keto-adapted). Still trying to find academic papers for the “epigenetics/transcriptomics/proteomics/chromatin structure/post-translational modifications” of ketosis (there’s indication that the alterations to chromatin structure could be longer-than-temporary if you do it for long enough)