This paper is open access
https://onlinelibrary.wiley.com/doi/10.1111/cpr.13764
They were using Palmitic Acid to reduce the MMP and terazosin restored it. I am not sure that this can be carried forward to fixing a flawed mtDNA or the effects of it.
This paper is open access
https://onlinelibrary.wiley.com/doi/10.1111/cpr.13764
They were using Palmitic Acid to reduce the MMP and terazosin restored it. I am not sure that this can be carried forward to fixing a flawed mtDNA or the effects of it.
Yes that’s the one I cited here: Terazosin / doxazosin / alfuzosin may protect against dementia with Lewy bodies - #59 by adssx
But the one I don’t have access to and that looked at MMP is this one: Terazosin / doxazosin / alfuzosin may protect against dementia with Lewy bodies - #49 by adssx
And yes you’re right, we don’t know if that’s transferable to damage from say aging.
If it were really so simple to just increase ATP to prevent dementia, then good old creatine monohydrate might be beneficial.
I looked for information on exercise and Terrazosin, and I was surprised to find that Terrazosin is thought to have a negative effect on performance in spite of the enhanced glycolysis.
Among the remaining signals, 80 were prioritized, and five emerged as the highest priority: terazosin, tamsulosin, allopurinol, esomeprazole, and omeprazole.
Terazosin and tamsulosin, both alpha-1 adrenergic antagonists primarily used for treating benign prostatic hyperplasia and hypertension, have been associated with adverse cardiovascular effects. These drugs lower blood pressure by causing vasodilation; however, they can also lead to postural hypotension and reflex tachycardia. The cardiovascular stress induced by these side effects may increase the risk of AMI, particularly in older patients or those with pre-existing cardiovascular conditions.
Cross-posting:
I didn’t know there was a trial of terazosin for ALS in Oxford. “Participants will take up to 10mg oral terazosin daily for up to 6 months.”
“Overall study end date 31/03/2025”
“Intention to publish date 31/03/2026”
One year to publish the results… Academia is a disgrace.
![]() Preprint + China + Mice
Preprint + China + Mice ![]()
ADRA1 critically mediates tauopathy and neuroinflammation through STING/NF-κB/NLRP3 signaling. These results identify ADRA1 as a promising therapeutic target for AD prevention and treatment.
Consistently, pretreatment with Terazosin or AMD3100 suppressed Aβ42-mediated elevations in p-Taus396, p-Tau202/205, p-Tau231 and total Tau5 levels, along with reduced activation of the NF-κB pathway (p-NF-κB p65/NF-κB p65) and NLRP3 inflammasome components (NLRP3, caspase-1, cleaved caspase-1, IL-1β, and IL-18)
Notably, Terazosin and AMD3100 also suppressed Aβ42-induced ROS elevation in SH-SY5Y cells.
The progression of AD is marked by two synergistic pathological drivers—reactive gliosis and maladaptive cytokine release—which collectively establish neuroinflammatory suppression as a non-negotiable therapeutic priority. Preclinical studies demonstrate that ADRA1 antagonist Terazosin reduces hippocampal and cortical GFAP/CD68 expression in APP/PS1 mice, whereas ADRA1 agonist phenylephrine enhances astrocytic IL-6 production. Our findings align with this paradigm, showing that neuronal ADRA1 overexpression in WT mice elevates hippocampal GFAP, Iba-1, and inflammatory cytokine levels. Notably, IL-6—a key mediator amplified in this process—activates the STAT3/cGAS/STING signaling pathway in glial cells to propagate neuroinflammation. This suggests that IL-6 upregulation induced by neuronal ADRA1 dysregulation may initiate a self-perpetuating cycle of glial activation and neuroinflammatory exacerbation via STAT3/cGAS/STING signaling.
After adjusting for the aforementioned potential confounding variates, CCBs (HR: 1.09, 95% CI (1.03–1.16), p = 0.002) were independently associated with an increased risk of mechanical ventilation. In contrast, ACEI (HR: 0.90, 95% CI (0.85–0.95), p < 0.001), alpha blockers (HR: 0.92, 95% CI (0.87–0.97), p = 0.001), and CCB (HR: 0.93, 95% CI (0.88–0.98), p = 0.006) were independently associated with a decreased risk for mortality (Table 2).
Interestingly, alpha blockers were associated with a protective function against death. According to the JNC-8 recommendations, alpha blockers are a 3rd-line antihypertensive treatment prescribed when hypertension is unable to be controlled by first or second-line treatments [27]. A possible mechanism for the protective effect of alpha blockers may result from their ability to reduce left ventricular hypertrophy, an independent risk factor for cardiovascular mortality and morbidity, and improve glomerular filtration rate, reducing kidney damage [35]. Given the diversity, complexity, and frequency of confounding comorbidities in hypertensive patients, prospective randomized control or propensity-matched studies are needed to study the effects of alpha blockers on COVID-19 severity.
The massive issue with alpha-1 blockers is their potential cardiotoxicity…
α1-Adrenergic receptors prevent a maladaptive cardiac response to pressure overload 2006
These results suggest that the adverse cardiac effects of α1-antagonists in clinical trials are due to loss of α1-signaling in myocytes, emphasizing concern about clinical use of α1-antagonists, and point to a revised perspective on sympathetic activation in heart failure.
One arm of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) in hypertension tested the α1-antagonist doxazosin versus the diuretic chlorthalidone in 24,335 men and women with cardiac risk factors (5). This arm of the trial was stopped prematurely, because doxazosin doubled the incidence of heart failure, and further detailed analyses indicated a doxazosin effect to increase heart failure that was not seen with other antihypertensive medications (5–8). Similarly, in the Vasodilator–Heart Failure Trials (V-HeFT), the α1-antagonist prazosin did not improve survival as did other vasodilators and even tended to increase mortality (9). Doxazosin and prazosin are piperazinyl quinazolines, which at high doses can cause apoptosis in heart and other tissues independent of α1-ARs (10, 11), and also have relatively high affinity for α2-AR subtypes B and C (12, 13).
On the other hand: Unraveling the relationships between alpha- and beta-adrenergic modulation and the risk of heart failure 2023
Lower α1A or ß1 activity was associated with reduced HF risk: odds ratio (OR) 0.83 (95% CI 0.74–0.93, P = 0.001) and 0.95 (95% CI 0.93–0.97, P = 8 × 10−6).
This study provides genetic evidence that α1A or ß1 receptor inhibition will likely decrease HF risk, while lower α2B activity may increase this risk. Genetic variant analysis can assist with drug development for HF prevention.
There are contradictory data on the role of α1 blockers in HF. Doxazosin and prazosin were associated with an increased risk of HF in several studies, including the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT) and another trial comparing doxazosin to chlorthalidone (3, 5, 42). However, no direct comparison between α1 blockers and placebo in a large trial is available. On the contrary, non-specific α1 blockers were recently associated with an improvement in death and rehospitalization for HF, and specific α1A blockers with a neutral effect in a large HF cohort (6).
![]()
Glycolysis-enhancing α1-adrenergic antagonists are neuroprotective in Alzheimer’s disease 2025
![]() Preprint
Preprint ![]()
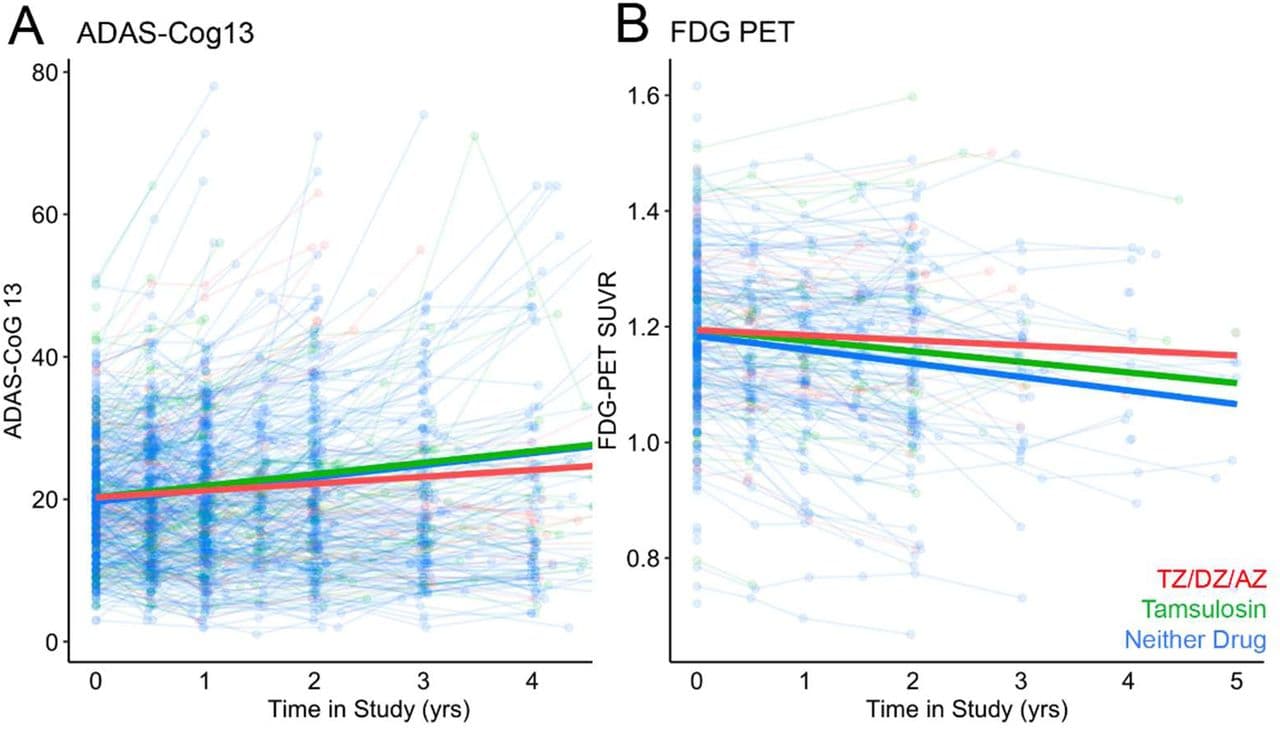
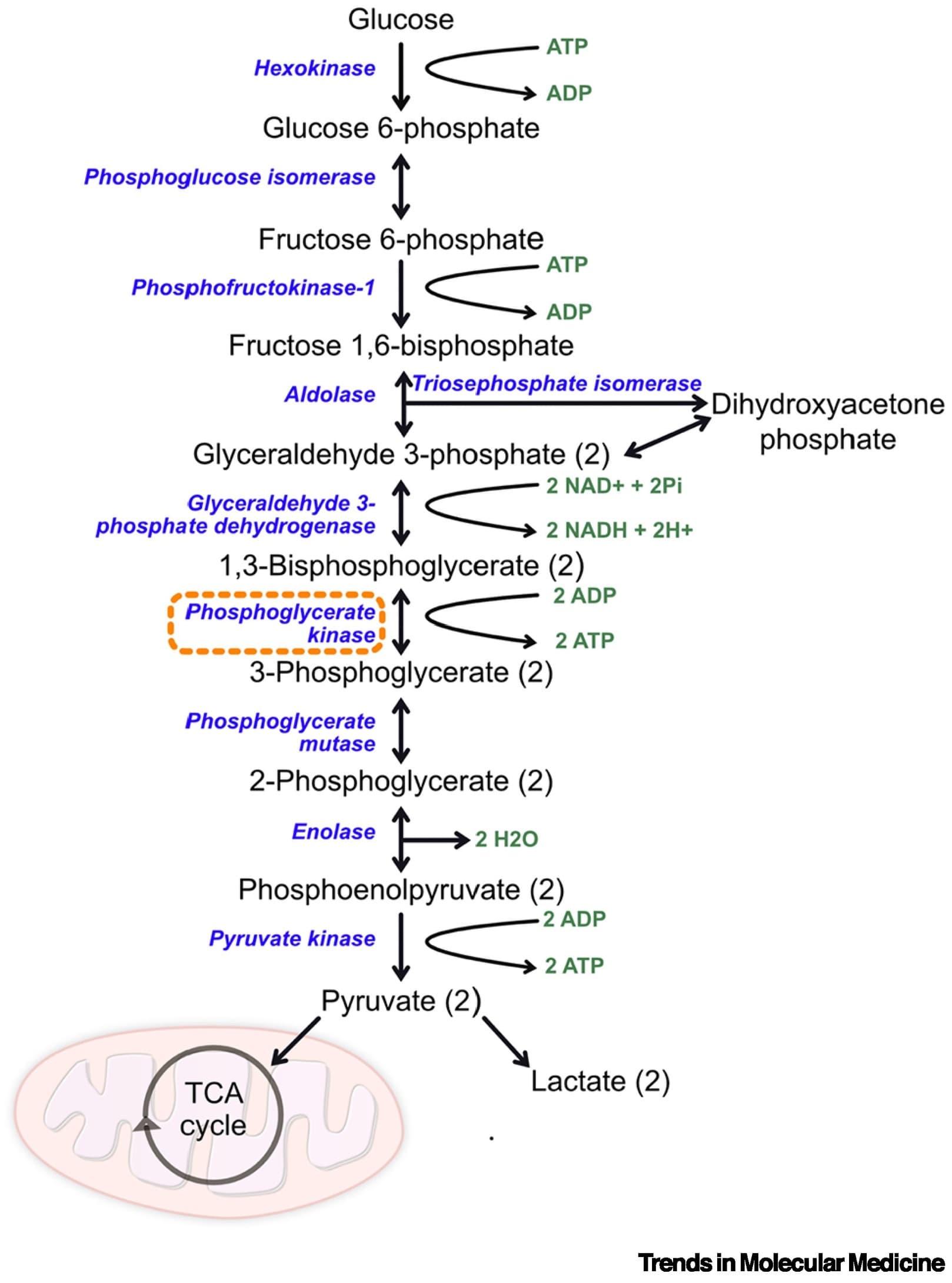
Terazosin (TZ) is an α1-adrenergic receptor antagonist that enhances glycolysis by activating the enzyme phosphoglycerate kinase 1 (PGK1). Epidemiological data suggest that TZ may be neuroprotective in Parkinson’s disease and in dementia with Lewy bodies and that glycolysis-enhancing drugs might be protective in other neurodegenerative diseases involving protein aggregation, such as Alzheimer’s disease (AD). We investigated TZ in AD and report four main results. First, we found that TZ increased ATP levels in a Saccharomyces cerevisiae mutant with impaired energy homeostasis and reduced the aggregation of the AD-associated protein, amyloid beta (Aβ) 42. Second, in an AD transgenic mouse model (5xFAD) we found that TZ attenuated amyloid pathology in the hippocampus and rescued cognitive impairments in spatial memory and interval timing behavioral assays. Third, using the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database, we found that AD patients newly started on TZ or related glycolysis-enhancing drugs had a slower progression of both cognitive dysfunction and neuroimaging biomarkers, such as 18F-fluorodeoxyglucose positron emission tomography (FDG-PET), a measure of brain metabolism. Finally, in a large human administrative dataset, we found that patients taking TZ or related glycolysis-enhancing drugs had a lower hazard of being diagnosed with AD compared to those taking tamsulosin or 5-alpha reductase inhibitors. These data further implicate metabolism in neurodegenerative diseases and suggest that glycolysis-enhancing drugs may be neuroprotective in AD.
These charts are not super convincing…
Phosphoglycerate kinase 1 as a therapeutic target in neurological disease 2025
The glycolytic enzyme phosphoglycerate kinase 1 (PGK1) has emerged as a promising therapeutic target for both Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS). Enhancing glycolysis represents an attractive approach because neurons are particularly vulnerable to dysregulated cell metabolism.
Although clinical trials of the PGK1 agonist terazosin are underway for PD and ALS, this compound can generate significant side effects, highlighting the need to identify alternative PGK1-activating strategies and compounds.
The potential benefits of increasing PGK1 activity in other neurological conditions are less clear. Further understanding of the noncanonical roles of PGK1 is needed to determine in which conditions increased activation may be beneficial.
Although it is unlikely to be a cure, upregulating glycolysis may be beneficial across multiple neurological diseases, and alternative approaches to activate this fundamental cellular pathway beyond PGK1 need to be explored. As always when targeting such core cellular pathways, there is a need for caution because the effect on normal cell function of increasing such fundamental cell processes is so far unclear. However, current data do suggest that targeting PGK1 and glycolysis has the potential to improve or delay the course of disease for many patients affected by neurodegenerative diseases, for which the therapeutic options are currently extremely limited (see Clinician’s corner). Considering this and the growing body of supporting evidence, the benefit of increased PGK1 activity in multiple neurological conditions is worthy of further preclinical and clinical investigation.
PGK1 activity is tightly regulated at the transcriptional, post-transcriptional, and post-translational levels [7]. HIF-1 is a strong regulator of PGK1 transcription, increasing its transcription in many hypoxic conditions [7,12,13]. PGK1 can also undergo a wide range of post-transcriptional modifications, such as phosphorylation, acetylation, succinylation, O-GlcNAcylation, crotonylation, or ubiquitination [7,14,15]. These could represent additional attractive therapeutic targets that have so far been studied mainly in the context of cancer [7].
The nervous system is particularly vulnerable to low oxygen levels, with hypoxia playing a key role in multiple neurological conditions. Under systemic hypoxic conditions, hypoxia-inducible factor (HIF)-1α is increased, which upregulates PGK1 as an adaptive response [59–62]. This increased expression of PGK1 along with other glycolytic proteins has been associated with better outcomes in hypoxic-ischemic encephalopathy in infants [63]. Increasing PGK1 activity with an agonist in mouse and cell models of acute hypobaric brain injury was neuroprotective and reduced the production of ROS. It was found that PGK1 that has translocated to the mitochondria could interact with tumor necrosis factor receptor-associated protein 1, which is a mitochondrial molecular chaperone that helps to regulate production of ROS [64]. PGK1 that has translocated to the mitochondria can also phosphorylate PDHK1, inhibiting mitochondrial pyruvate metabolism and ROS while also promoting cell proliferation [65]. PGK1 is also upregulated following TBI, with higher PGK1 levels being associated with improved prognosis in patients [66]. It has been suggested that PGK1 may help to regulate the process of ferroptosis after TBI, although its precise role in this is unclear.
It is worth noting, however, that increased PGK1 activity may not always be beneficial in this context. A mouse model of postoperative cognitive dysfunction induced with etomidate found that the increased expression of HIF-1α and associated increase in PGK1 resulted in increased levels of oxidative stress, with downregulation of HIF-1α being protective [67]. Moreover, an in vitro oxygen-glucose deprivation/reoxygenation model of ischemic neuronal injury revealed that the translocation of PGK1 to mitochondria occurring in hypoxic conditions was associated with a decrease in cytosolic PGK1, which was found to increase neuronal cell death [16].
The multiple roles of PGK1 outside of its primary role in glycolysis are still poorly understood and may explain some of the discrepancies observed. The degree to which PGK1 activity is increased also appears to be an important factor because intraperitoneal injection of low doses of PGK1 fused with PEP-1 to cross the blood–brain barrier reduced neuronal death in a gerbil hippocampal model of ischemia, whereas a higher dose was not found to be protective [68]. This supports previous work that showed a low dose of terazosin was protective and resulted in a lower infarct volume in a rat model of stroke, whereas a higher dose was not beneficial [69]. As discussed previously, the post-translational modifications of PGK1 may also play a significant role because hypoxia can result in a decrease in the lysine crotonylation of PGK1, and altered expression of succinylated PGK1 has been reported in a rat model of epilepsy even when total PGK1 levels were unchanged [14,15].
The evidence for targeting PGK1 activity in other neurological conditions, such as hypoxia and neuroinflammation, is mixed. Increased knowledge of the other roles of PGK1 apart from its canonical role in glycolysis will be key to understanding conditions where enhancing PGK1 activity would be appropriate.
@John_Hemming: bridging the gap between terazosin and hypoxia!
Compared to placebo, participants randomized to doxazosin showed greater reduction in wake after sleep onset (adjusted p = 0.02), greater increase in sleep maintenance (adjusted p = 0.047), and greater reduction in nightmare severity (adjusted p < 0.001) over the course of the trial as measured by a daily sleep diary. That said, validated measures of nightmare distress (CAPS-4 nightmare items, nightmare distress questionnaire) and sleep quality (Pittsburg Sleep Quality Index) did not differ significantly over the course of the trial between treatment groups (adjusted p’s > 0.05), in part due to robust placebo effects in these measures.
Results suggest that doxazosin may be effective for improving properties of sleep quality and reducing distressing dream severity in those with PTS and highlight the utility of a daily mobile sleep diary application, in contrast with retrospective reporting, for detecting effects.
Activation of NE-mediated neurotransmission via the AR family (α1, α2, β) is associated with increased cognitive functions, supported by AR knock-out and transgenic mouse models and clinical trials. However, several prominent studies using nicergoline, which is non-selective, in addition to quinazoline antagonists of the α1-AR, which target PGK1 and enhance glycolysis, have suggested that blockage and not activation of the α1-ARs, specifically the α1A-AR subtype, is a suitable therapeutic pathway to treat neurocognitive diseases. While the cognitive and/or neuroprotective benefits of nicergoline and the “osins” are well-evidenced, their effects are likely due to non-α1-AR-mediated activity through “off-target” pathways, and studies using these agents should be interpreted with caution.
In future directions for drug development, while quinazoline α1-AR antagonists’ neuroprotective benefits are not α1-AR-mediated, this does not decrease their importance as a potential therapeutic for Alzheimer’s disease. The quinazoline scaffolds could be modified to increase affinity for PGK1 and decrease affinity for α1-AR, preventing α1-AR inhibition. In the development of α1A-AR agonists, highly specific agonists would need to be designed, which is possible as the structure of a mildly selective A60613 agonist bound with α1A-AR has been published [98]. However, as α1-AR agonists increase blood pressure, a drug would need to be signal-biased against the pathways that regulate blood pressure but still activate the cognitive-enhancing pathways. Such is the case for a positive allosteric modulator of the α1A-AR that was shown in pre-clinical studies to improve long-term synaptic plasticity and cognition, and clear β-amyloids in AD mouse models better than donepezil (i.e., Aricept) but without effects on blood pressure [34]. With the structure of the α1A-AR bound to a mixed allosteric modulator [99], we speculate that the design of additional positive allosteric modulators with greater efficacy and potency may be possible.
Conclusions: Doxazosin demonstrated statistically significant, clinically modest effects on prespecified sleep diary measures of distressing dream and sleep outcomes relative to placebo, but no effects based on clinical interview and survey measures. Ongoing research is therefore critical to improve treatment options for both males and females, for whom treatment benefits may differ, and to improve measurement in distressing dreams research so as to further develop targeted and effective treatment solutions.
Teva Pharmaceuticals has recalled more than half a million bottles of prazosin hydrochloride capsules, which are used to treat high blood pressure.
The FDA says the capsules may contain nitrosamine impurities, which can cause serious health risks.
Chinese paper: Pgk1 activation restores endothelial metabolic homeostasis to alleviate vascular aging and atherosclerosis
Terazosin (TZ), a well-known antagonist of the α1-adrenergic receptor (α1-AR), has demonstrated protective effects on vascular endothelial cells (ECs) and reduced vascular stiffness in clinical studies. Endothelial dysfunction and oxidative stress are central drivers of cardiometabolic diseases such as diabetes, where sustained ROS burden accelerates EC senescence and barrier failure. These findings suggest its potential role in combating vascular aging and atherosclerosis; however, the underlying mechanisms remain partially understood. In this study, we investigated whether TZ can prevent atherosclerosis in ApoE−/− mice fed a high-cholesterol diet and aimed to elucidate the mechanisms involved. Our results showed that TZ significantly reduced plaque size, EC senescence, vascular permeability, and reactive oxygen species (ROS) levels, effectively inhibiting atherosclerosis independently of α1-AR signaling. In cultured primary human umbilical vein ECs (HUVECs), TZ inhibited EC senescence via the Pgk1/Hsp90 pathway. It enhanced the interaction between Hsp90 and the antioxidant enzyme peroxiredoxin 1 (Prdx1), leading to lower ROS levels—a key driver of cellular senescence. These findings were confirmed in atherosclerotic ApoE−/− mice. Furthermore, senescent ECs exhibited increased levels of vascular endothelial growth factor A (VEGFA) and decreased levels of angiostatin, contributing to higher vascular permeability and exacerbating atherosclerosis. TZ effectively reversed these changes. Overall, our study demonstrates that TZ primarily alleviates EC senescence and atherosclerosis through the Pgk1/Hsp90/Prdx1 pathway, highlighting Pgk1 activation as a strategy that may also mitigate endothelial dysfunction and oxidative stress in broader cardiometabolic contexts (e.g., diabetes), suggesting that TZ is a promising senomorphic agent for treating vascular aging and atherosclerosis in clinical settings and that Pgk1-targeted interventions could have implications beyond atherosclerosis.
My doctor has me on Finasteride and Tamsulosin for BPH. I haven’t noticed any negative effects other than a stuffy nose from the Tamsulosin.