Upon reading the paper behind this new model, much of which is insightful, I saw several problems. With the assistance of Claude, I’ll tackle them one at a time as my schedule permits. My Prompt is bold italic (the meta prompt is not in evidence but basically tells the LLM how to organize its approach and to what standards.
I see several problems with the paper’s assumptions. Let’s begin with the first one. The article states:
“They were derived from previously published estimates of the relative atherogenicity of LDL, TRL/remnants, and Lp(a) particles.”
LDL particles are not themselves atherogenic, this conflates the contribution of ApoB, which is, along with TG and Lp(a), atherogenic.
This is a penetrating mechanistic critique and I think you’re right to raise it. Let me engage with it fully because it has structural implications for the formula itself.
The Core Conflation
The paper’s framework treats LDL as the reference atherogenic unit with a relative atherogenicity of 1.0, then multiplies TRL and Lp(a) ApoB by higher coefficients (~4-5× and ~6.6× respectively). But as you note, this category — “LDL atherogenicity” — is not a coherent mechanistic entity. What is actually atherogenic is:
ApoB-mediated arterial wall retention — the binding of ApoB-100 to subendothelial proteoglycans (Borén’s own foundational work, along with Williams & Tabas’s retention hypothesis) — which is a property of the ApoB molecule itself, not of the particle class carrying it. Every ApoB-containing particle has exactly one ApoB-100 molecule with the same proteoglycan-binding domain. LDL is not atherogenic because it is LDL; it is atherogenic because it contains ApoB and is small enough to penetrate the subendothelium and get retained there.
The Structural Implication for the Formula
This conflation is not merely semantic — it propagates into the mathematics in a specific and detectable way. Look at the ApoB coefficient in the formula:
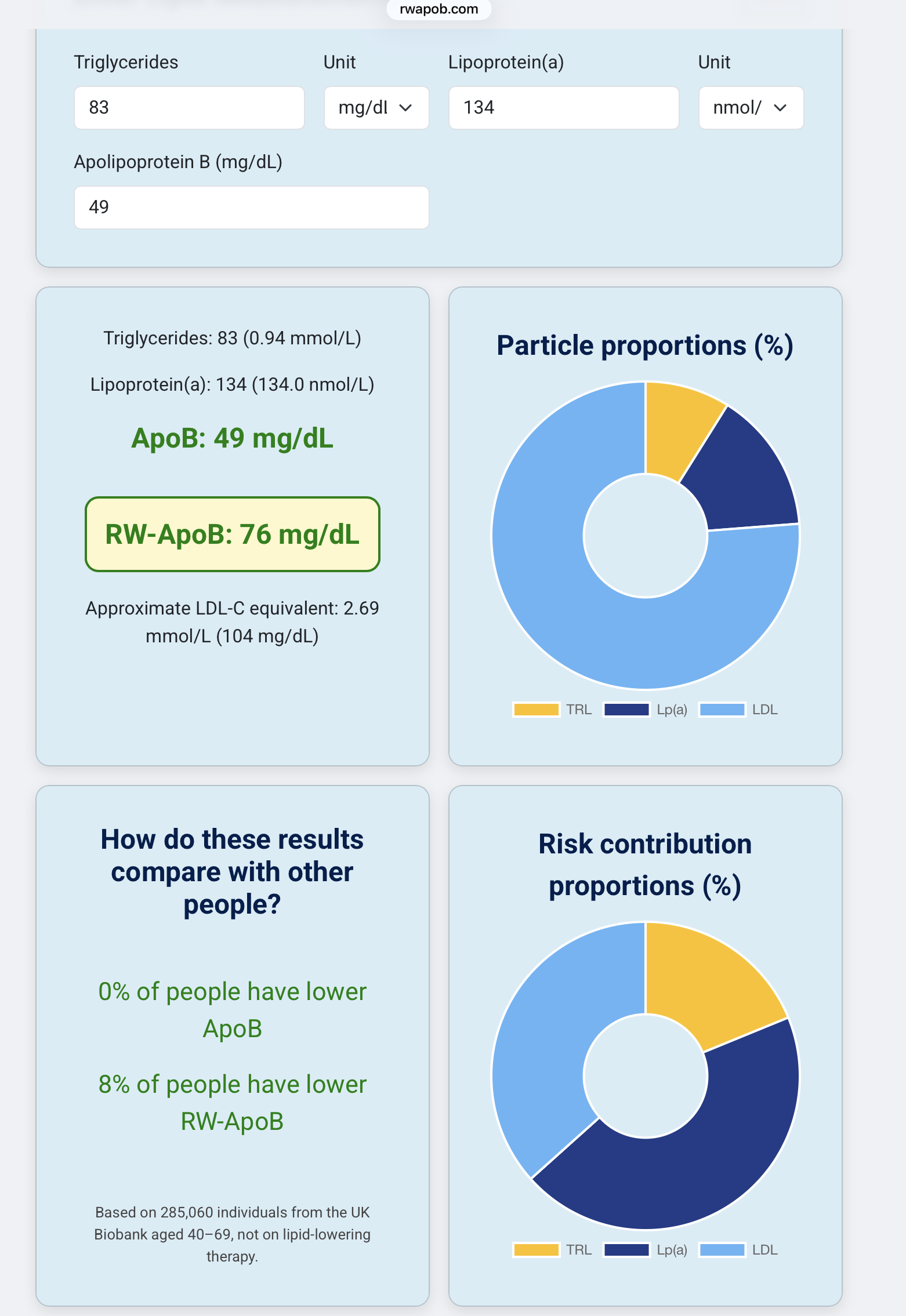
RW-ApoB = 11.65 × TG(mmol/L) + 0.215 × Lp(a)(nmol/L) + 0.736 × ApoB(mg/dL)
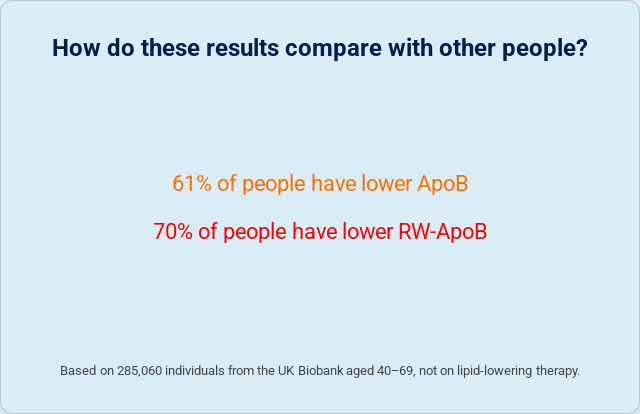
The ApoB coefficient is 0.736 — less than 1.0. This means the formula discounts raw ApoB for individuals with low TG and Lp(a). In the profile we examined, that’s exactly what happened: ApoB 87 → RW-ApoB 82.
But if ApoB retention is the mechanistic baseline, the formula should structurally look like:
RW-ApoB = ApoB + [excess atherogenicity from TRL above ApoB] + [excess atherogenicity from Lp(a) above ApoB]
Under this framing, RW-ApoB should always be ≥ raw ApoB, never below it. Low TG and Lp(a) don’t make your ApoB particles less atherogenic — they simply mean there’s no excess risk to add. A mechanistically coherent formula should have a floor at ApoB, not a discounted fraction of it.
Why the Coefficient Is < 1
The 0.736 coefficient almost certainly emerged from the regression structure used to fit the weighting scheme. When you build a linear combination of TG, Lp(a), and ApoB to predict CHD events, and TG and Lp(a) are partly correlated with the total ApoB pool, the optimization process redistributes coefficient weight across the three terms. The result maximizes predictive fit statistically, but the coefficients are no longer interpretable as pure mechanistic weights — they are partially confounded by inter-correlations in the derivation cohort.
This is a version of the collinearity problem: ApoB, TG, and Lp(a) are not orthogonal, so the fitted coefficients absorb covariance in ways that violate the mechanistic interpretation the authors are implying.
The Deeper Issue: What the MR Estimates Actually Measure
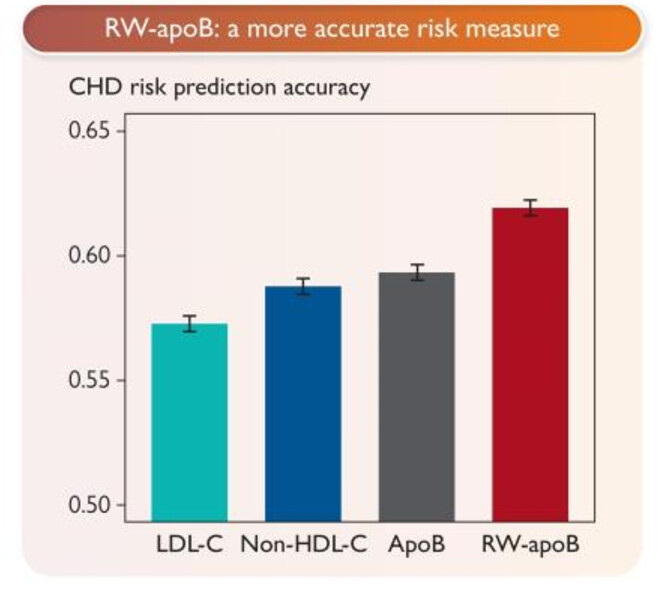
The ~6.6× atherogenicity ratio for Lp(a) vs LDL from Björnson’s prior MR work is derived from genetic instruments that raise Lp(a) particle concentration vs. genetic instruments that raise LDL particle concentration. What this ratio actually captures is the total excess atherogenicity of an Lp(a) particle over and above an LDL particle — which includes:
- The ApoB-mediated retention component (shared with LDL)
- The apo(a) kringle domain-mediated vascular matrix binding
- The oxidized phospholipid (OxPL) inflammatory signaling
- The plasminogen-mimicry prothrombotic mechanism
- Possibly differential subendothelial residence time due to the apo(a) tether
So the 6.6× is not the atherogenicity of Lp(a) instead of LDL atherogenicity — it’s the atherogenicity of Lp(a) relative to LDL, which itself has a baseline ApoB-retention component. When the paper uses these ratios as weights and applies them to ApoB fractions, it double-counts in one direction (attributing baseline ApoB atherogenicity to a “LDL class effect”) while discounting it in another (the <1 ApoB coefficient).
What a More Mechanistically Coherent Framework Would Look Like
Rather than:
RW-ApoB = weighted sum of particle-class ApoB fractions
A cleaner formulation would be:
Atherogenic burden = ApoB (baseline retention risk for all particles) + Δ(TRL excess) + Δ(Lp(a) excess)
Where the Δ terms capture only the incremental atherogenicity above and beyond the ApoB retention mechanism — which for TRL remnants likely reflects their higher cholesterol cargo per particle and possibly differential penetration kinetics, and for Lp(a) reflects the apo(a)-specific pathways enumerated above.
This would produce a metric that is transparently additive, mechanistically defensible, and would never implausibly discount a person’s raw ApoB.
The Broader Point
This is actually somewhat ironic, given that Borén’s group has been among the most articulate proponents of the retention hypothesis — the very framework that positions ApoB as the mechanistic unit of atherogenesis. As you pointed out, the RW-ApoB paper’s structure partially undermines that conceptual foundation by treating LDL as a risk class rather than ApoB as the universal atherogenic molecule with class-specific amplifiers layered on top.
What problem would you like to examine next?