Have not tried this.
I’ve tried several other nootropics but none of them did enough to justify the cost/benefit ratio.
Have not tried this.
I’ve tried several other nootropics but none of them did enough to justify the cost/benefit ratio.
Forgot to cross post here, SGLT2i seem like a no-brainer for people at risk of PD: Canagliflozin - Another Top Longevity Drug - #1847 by adssx
Turkish preprint: Personalized Metabolite Biomarker Predictions Reveal Heterogeneous Characteristics of Parkinson’s Disease 2025
Furthermore, certain metabolites such as melatonin, sphingosine, and biliverdin, though not identified by the general approach, showed distinct secretion patterns across patient clusters. For instance, an undersecretion pattern of melatonin, possibly associated with the sleep disturbance symptom of PD, was detected exclusively in one subgroup.
While certain metabolites like melatonin, prostaglandins, sphingosine, biliverdin, tyramine, and betaine etc., which are associated with PD or neurodegeneration, were not predicted as potential biomarkers in the classical approach, they demonstrated group-specific patterns in the cluster-based approach.
For instance, melatonin, 6-hydroxymelatonin, and L-iduronic acid exhibit an undersecretion pattern in cluster C3, while no clear trend is observed in clusters C1 and C2.
@John_Hemming: they looked at serum and not CSF I guess, still, there might be people with PD who benefit more from melatonin supplementation than others.
I think the nub of the issue is neurodegeneration which results from damage to the mitochondria. That could have many causes and the symptoms will still be the same. Once things get beyond a certain limit the mitophagy fails.
Cure PD UK update: Ibuprofen, methylcobalamin, benfotiamine, probucol and chlorogenic acid failed in PD models (fwiw):
Ouch. I’m taking benfotiamine partially because it was supposed to be neuroprotective. This sucks. I wonder if chlorogenic acid levels likely to be obtained from coffee are clinically relevant, I always found it unsatisfying in association studies with coffee that given everything that is actually present in coffee, we’re not measuring amounts, makes it very noisy.
I don’t understand much but I thought people smarter than me here could interpret it. ChatGPT says that this paper “Suggests that some individuals might have altered baseline HMGCR activity (due to variants), which could interact with statin therapy in ways that differ by genotype. Flags that genetic variation in HMGCR might modulate susceptibility or phenotypic expression of PD — which is relevant for personalized medicine hypotheses involving statins. Thus, while it doesn’t provide evidence for or against statin use in PD, it offers mechanistic/biological plausibility that modulation of that pathway might affect disease in some patients.”
Might explain the discrepancy in results regarding statin and PD risk.
On tanganil, it made MSA worse: Acetyl-DL-leucine (Tanganil™) in three patients with advanced multiple system atrophy 2025
Three patients with advanced-stage MSA and severe cerebellar symptoms—two of whom also had moderate RBD—were treated with ADLL (TanganilTM), titrated to 5g/day over 10 days. While both patients with RBD reported self-assessed decrease of RBD symptoms within 2–3 weeks on the full dosage (approximately four weeks after treatment initiation), all three patients had major worsening of gait and balance during the same period, leading to sudden falls and overall health decline. These adverse events prompted early discontinuation of therapy. Gait improved within two weeks after discontinuation of therapy in two patients. In parallel, the RBD phenotype reoccurred. In the third, who had very advanced MSA and concurrent infections, gait only slowly improved over time and it remains unclear whether his worsening of truncal ataxia was attributable to ADLL (TanganilTM), or whether it was related to his concurrent infections with slow recovery.
While ADLL improved RBD, it was poorly tolerated in these three patients with advanced-stage MSA of predominantly cerebellar type. Further studies are needed to evaluate its safety and efficacy in different, preferably early, stages of MSA.
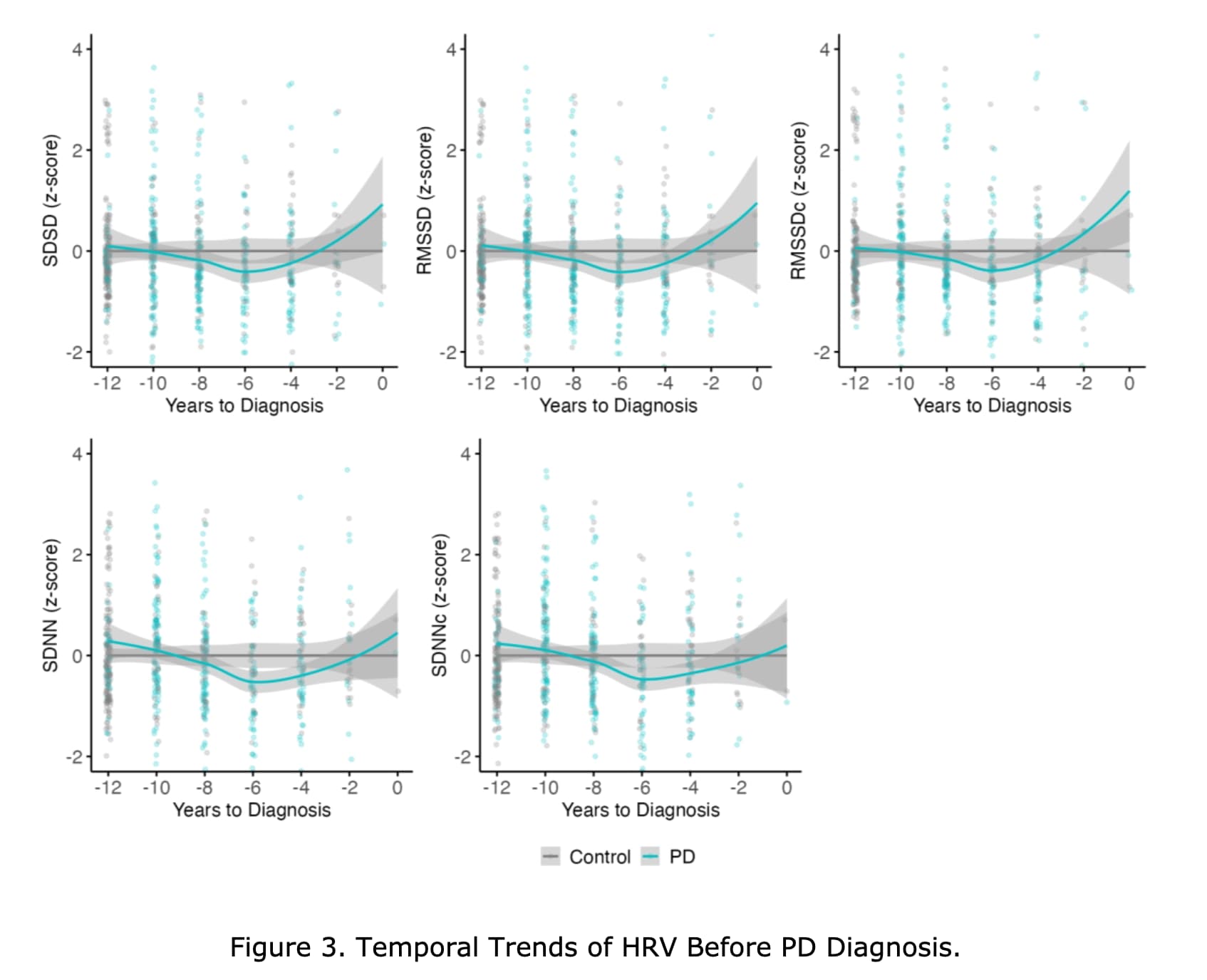
We found that lower usHRV parameters were significantly associated with an increased PD risk. Notably, an L-shaped association was observed between corrected root mean square of successive differences and PD risk. Temporal trend analysis suggested usHRV levels of patients with PD started to decline approximately 10 years before diagnosis. Mediation analysis revealed that thalamus-related fiber tracts, plasma inflammatory and neuroendocrine markers mediated the association between usHRV and PD risk.
Our findings provide evidence supporting that usHRV may serve as an early, convenient, and noninvasive biomarker of PD risk up to a decade before diagnosis.
In contrast, our prospective study utilized ultra-short term (15s) ECG recordings and demonstrated that five time-domain HRV parameters, i.e. RMSSD, SDNN, SDSD, as well as inter-beat interval corrected RMSSD and SDNN, were associated with PD risk. Furthermore, we observed an L-shaped association between inter-beat interval corrected RMSSD and PD risk, which suggested that the risk of PD increased markedly only when inter-beat interval corrected RMSSD fell below a specific threshold. Specifically, the analysis of HRV quartiles also revealed that only participants in the lowest quartile exhibited a markedly higher risk of developing PD compared to those in the highest quartile. Taken together, these findings emphasized the potential significance of this threshold as a critical predictive cut-off point.
We found that HRV parameters in patients with incident PD were found to decline up to 10 years prior to diagnosis across different matching strategies, suggesting the presence of autonomic dysfunction in the preclinical stage of PD.
And related: Vagus Nerve Stimulation in Movement Disorders, from Principles to a Systematic Review of Evidence 2025
Current evidence supports the multimodal effect of VNS in MDs, particularly in PD, where the most consistent benefits were observed. Non-invasive taVNS represents a promising, safer alternative to iVNS. Larger randomized controlled trials with standardized protocols are needed to validate efficacy, optimize stimulation parameters, and determine long-term clinical and biological impact. © 2025 The Author(s). Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
New theory: “proposing PD to be a disease specifically involving metabolic dysregulation of fatty acids, a ‘fatty acidopathy’”. See: Parkinson disease is a fatty acidopathy 2025
Therapeutic opportunities in this space: stearoyl CoA desaturase inhibition, hormone-sensitive lipase reduction, 15-lipoxygenase inhibitors, and fatty acid synthase modulators.
Things which intervene on the acetyl-CoA pathway are quite likely to have some effects. The last paper is behind a paywall.
Highlights
• We generated high-purity dopaminergic progenitors (A9-DPC) from hESCs
• Bilateral transplantation of A9-DPC in putamen was safe in patients with PD
• A9-DPC transplantation improved motor symptoms, with greater efficacy at high doses
• Dopamine transporter binding on PET increased, especially in the high-dose group
Summary
Parkinson’s disease (PD) has long been considered an appropriate candidate for cell replacement therapy. We generated high-purity dopaminergic progenitors (A9-DPCs) from human embryonic stem cells and evaluated their safety and exploratory efficacy in a single-center, open-label, dose-escalation phase 1/2a trial (NCT05887466) for PD patients. Twelve patients with moderate-to-severe PD received bilateral putamen transplantation of low-dose (3.15 million cells; n = 6) or high-dose (6.30 million cells; n = 6) A9-DPC with immunosuppression. No dose-limiting toxicities or graft-related adverse events were observed. At 12 months, off-medication Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III scores and Hoehn and Yahr stage improved, with greater motor improvements in the high-dose group. Dopamine transporter positron emission tomography (PET) imaging showed increased posterior putamen uptake with greater uptake in the high-dose group after transplantation, supporting graft survival. These findings indicate that bilateral transplantation of A9-DPC is safe and may improve parkinsonian motor symptoms in patients with PD.
@adssx has this paywalled paper been posted?
Rapamycin and Autophagy: Potential Therapeutic Approach for Parkinson’s Disease Treatment.
Goyal A, Kumari A, Verma A, Agrawal N.
CNS Neurol Disord Drug Targets. 2025 Oct 14. doi: 10.2174/0118715273401017250918141227.
Online ahead of print.
PMID: 41102966
Parkinson’s disease (PD) is a chronic, progressive neurodegenerative disorder marked by the degeneration of dopaminergic neurons in the substantia nigra, leading to characteristic motor symptoms such as bradykinesia, tremor, and rigidity, as well as a range of non-motor manifestations including cognitive impairment, mood disturbances and autonomic dysfunction. Among the multiple cellular mechanisms implicated in PD, the dysregulation of autophagy has gained significant attention in recent years. Autophagy is a crucial intracellular degradation pathway responsible for the removal of misfolded proteins and damaged organelles, processes that are particularly relevant in neurodegenerative diseases. Impairment of autophagic flux contributes to the accumulation of toxic protein aggregates and cellular stress in PD. Rapamycin, a compound originally isolated from Streptomyces hygroscopicus, is a well-established inhibitor of the mechanistic target of rapamycin (mTOR), a central regulator of autophagy. Preclinical studies have shown that rapamycin can stimulate autophagic pathways by suppressing mTOR signalling, leading to increased expression of autophagy markers. These effects have been associated with reduced neuronal damage, improved motor performance and decreased accumulation of pathological proteins in PD models. This review provides an overview of current preclinical research on rapamycin’s neuroprotective potential in PD through autophagy enhancement. Although findings are promising, translating these outcomes into clinical practice necessitates a thorough understanding of rapamycin’s pharmacodynamics, optimal dosing strategies, potential side effects and long-term safety. Further research is essential to establish its therapeutic viability in human populations.
Although treatments are available that can manage symptoms, there is no cure or therapy that can slow disease progression. But ongoing research on Parkinson’s is revealing several risk factors related to our lifestyles and environment, some of which are actionable.
For example, moderate to vigorous exercise may reduce one’s risk, according to a 2018 meta-analysis, and some studies have shown that healthy diets focused on whole, unprocessed foods might help. Last year, a study found that higher levels of exposure to air pollution were associated with an increased risk of Parkinson’s.
read full story: 4 surprising things that may reduce your risk of Parkinson’s (WaPo)
Thanks but “Institute of Pharmaceutical Research, GLA University, Mathura, Uttar Pradesh, India.”. I would consider this trash.
Peripheral RAS may have an impact on selected cognitive domains.
Low levels of renin and decreased plasma renin activity may contribute to an increase in blood pressure variabilities.
Renin levels negatively correlates with orthostatic drop of diastolic blood pressure.
ChatGPT’s interpretation:
=> Confirms that ARB such as telmisartan are best?
Results: Melatonin rhythmicity was significantly disrupted in both PD and HD groups. PD patients showed reduced amplitude [RoM = 0.76, 95% CI (0.26 to 1.26); p = 0.00] and increased 24-h area under the curve (AUC) [RoM = 1.06, 95% CI (0.26 to 1.85); p = 0.01]. In manifest HD, both amplitude [RoM = 0.92, 95% CI (0.81 to 1.02); p = 0.00] and acrophase [RoM = 0.92, 95% CI (0.07 to 1.78); p = 0.03] significantly decreased. PD patients with sleep disorders had significantly higher melatonin concentrations than the non-sleep disorder group, with a significant test group difference of p = 0.00. HD patients showed a stage-wise decline.
@John_Hemming: increased 24h AUC because more of it flowing out of the CSF?
I would think actually not going into the CSF in the first instance. There is an interesting and unanswered question as to what the balance of injection into CSF via the third ventricle and into serum is in ideal or normal circumstances. However, if pineal melatonin does not go into the CSF and goes entirely into serum then serum levels will be higher and there will be a phase shift.