@John_Hemming’s theory vindicated? Just published: Citric acid disassembles α-synuclein fibrils and reduces their cytotoxicity 2025
![]() Korean paper from an okay but not Tier 1 institution + Short publication and not long paper + Mechanistic in vivo model
Korean paper from an okay but not Tier 1 institution + Short publication and not long paper + Mechanistic in vivo model ![]()
Misfolding and aggregation of α-synuclein are associated with the progression of Synucleinopathies including Parkinson’s disease
Citric acid can disrupt the β-sheet structure of α-synuclein fibrils and inhibit additional fibril formation
The molecular docking simulation shows that citric acid exhibits a strong affinity for β-sheet stacking and adjacent regions
Citric acid reduces the cytotoxicity of α-synuclein fibrils
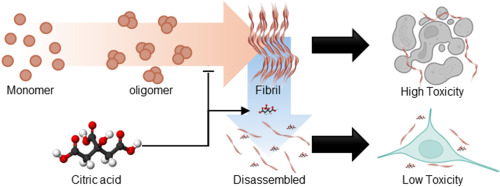
α-synuclein plays a crucial role in regulating neurotransmitter release and synaptic plasticity [1]. However, the misfolding and aggregation of α-synuclein are major hallmarks of Lewy bodies related to progressive neurodegenerative disorders, including Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy [2]. Thus, developing therapeutics that target α-synuclein aggregates is crucial for treating synucleinopathies, as it alleviates neurotoxicity [3]. Herein, we demonstrate that citric acid (citrate, 2-hydroxy-propane-1,2,3-tricarboxylic acid), commonly found in food supplements, can disassemble α-synuclein fibrils and mitigate their neuronal toxicity (Fig. 1A). Our findings reveal that citric acid can effectively disrupt β-sheet structure of α-synuclein fibrils and inhibit further fibril formation. Molecular docking (MD) simulation further supports these results, showing that citric acid exhibits a high affinity for the β-strand and the surrounding region within α-synuclein fibrils. We propose that citric acid holds significant promise for pharmaceutical applications in treating neurodegenerative diseases related to synucleinopathies.
In summary, we demonstrated that citric acid can effectively inhibit the α-synuclein fibril formation and disaggregates existing fibrillar aggregates. It also reduces the cytotoxicity of α-synuclein fibrils and interacts with the β-strand regions and their surrounding residues within the fibril structure. Given that pathological α-synuclein aggregates may originate in the gut, orally administered citric acid could act locally to disassemble them before brain propagation. Notably, although typical plasma citrate levels are around 0.1 mM, gastrointestinal concentrations can transiently reach 10–20 mM after citrus ingestion, supporting the feasibility of our effective dose for gut-targeted intervention.