Mouse model and “Parkinson-like” (not PD itself). Copper intake is not associated with a lower risk of PD: Dietary intake of iron, zinc, copper, and risk of Parkinson’s disease: a meta-analysis 2015
2 Likes
6. Bottom line
- Yes, aberrant splicing can mis-fold SOD1 and even present clinically with Parkinsonism—but so far this looks like a medical curiosity, not the rule.
- In mainstream idiopathic PD, all available evidence points to metal-binding and other post-translational disturbances, not splicing, as the proximate cause of SOD1 mis-folding.
That balance of evidence should guide both mechanistic thinking and therapeutic prioritisation.
Whichever way, however, SOD1 errors will cause further mitochondrial damage.
2 Likes
Yes: mice, and “PD-like”. That’s obviously limitations.
But from the paper I posted:
“We evaluated whether the blood-brain-barrier-permeable copper delivery drug, CuATSM, attenuated the misfolding and deposition of wild-type disSOD1 and associated neuron death in a novel mouse model that expresses this pathology.”
The paper you posted refers to dietary intake of copper. These may not be the same effect, quite apart from the form or delivery mechanism - this might also be an effect of administering a bolus to affect the relevant tissue. In other words, very different from dietary intake effects. YMMV.
1 Like
[!]HEAVY CAVEATS[!]: imrpress India study; in mice; MPTP-induced PD.
Telmisartan Protects Mitochondrial Function, Gait, and Neuronal Apoptosis by Activating the Akt/GSK3β/PGC1α Pathway in an MPTP-Induced Mouse Model of Parkinson’s Disease
3 Likes
Telmisartan is very promising. There is an ongoing RCT in the UK.
4 Likes
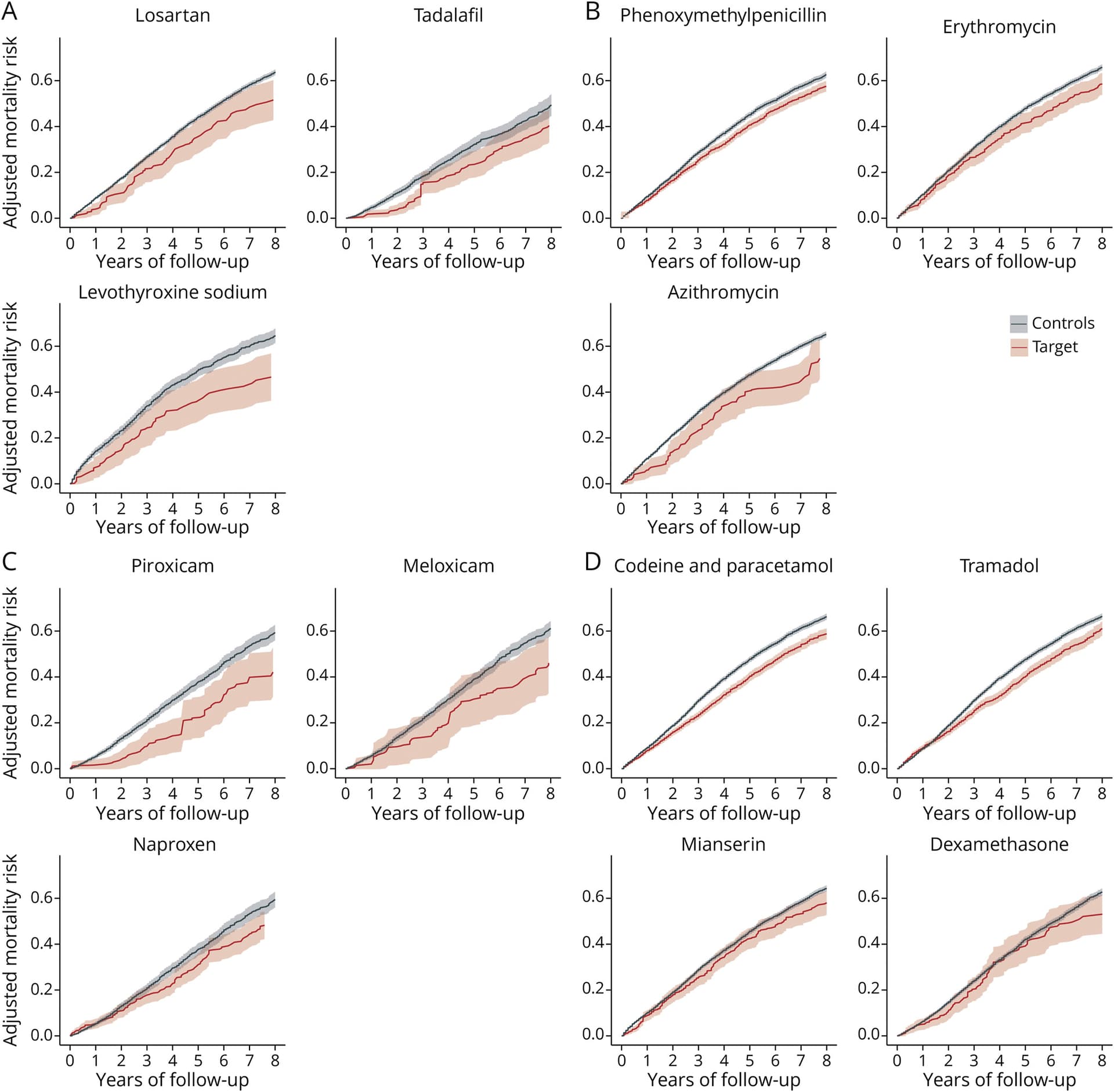
Very good paper: Association of Medication Use and 8-Year Mortality Risk in Patients With Parkinson Disease: Drug-Wide Trial Emulation 2025
The study included 14,289 individuals with PD (mean age 72 at diagnosis, 59% male) and identified 23 drugs associated with reduced mortality risk at 8 years. These drugs included ranitidine (histamine-2 blocker); pantoprazole and esomeprazole (proton pump inhibitors); losartan (angiotensin receptor blocker); atorvastatin (for high cholesterol); tadalafil (for erectile dysfunction); levothyroxine sodium (thyroid hormone); phenoxymethylpenicillin, erythromycin, and azithromycin (antibiotics); 4 nonsteroidal anti-inflammatory drugs; combined codeine/paracetamol and tramadol (opioid analgesics); zopiclone and melatonin (sleep aids); mianserin (antidepressant); mometasone (nasal corticosteroid); 2 opium-derived cough medicines; and dexamethasone (ophthalmologic corticosteroid).
The four NSAIDs are piroxicam, meloxicam, naproxen, and glucosamin and the two cough medicines are ethylmorphine with and without a combined expectorant.
Poke @John_Hemming for melatonin of course ![]()
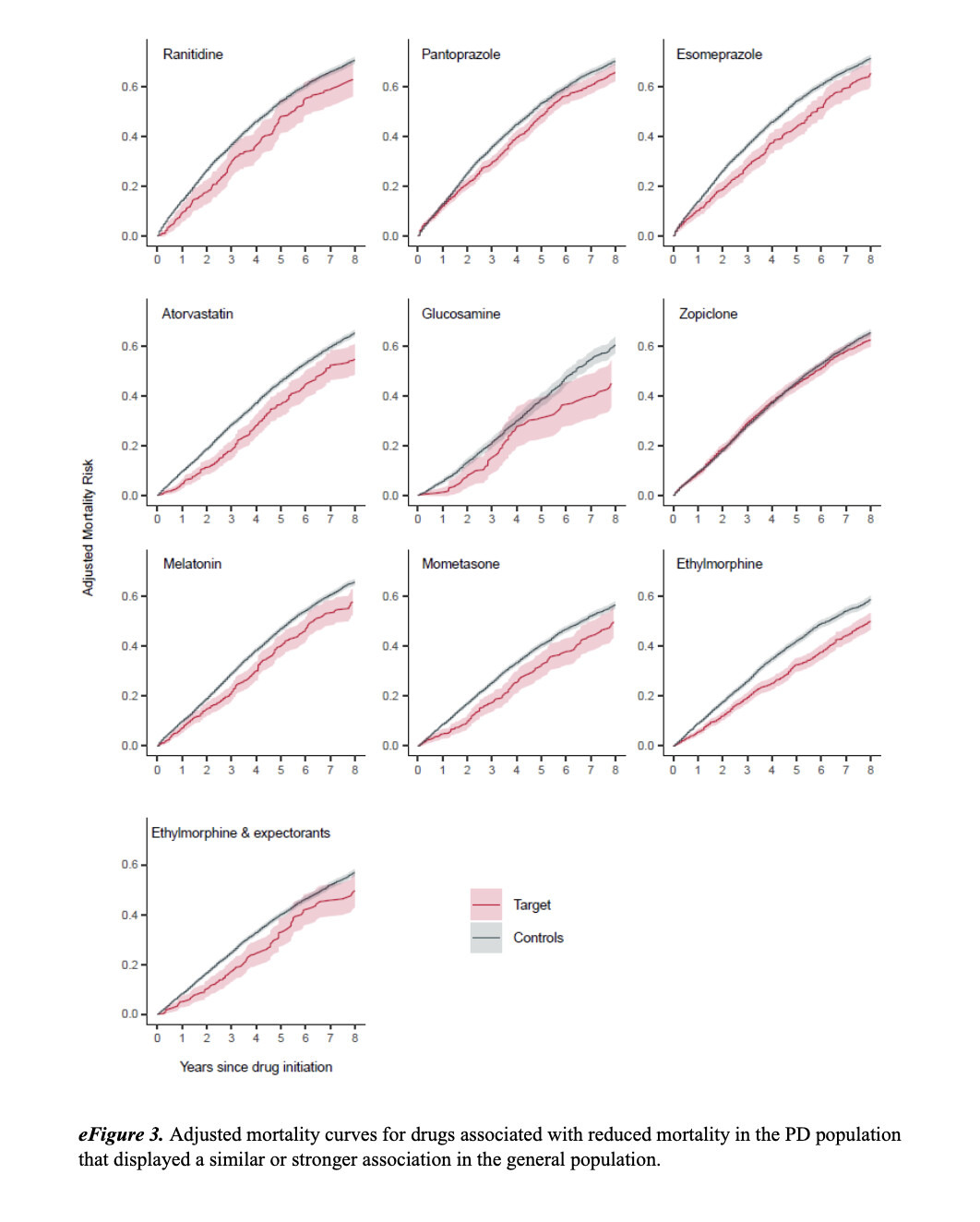
Our study also has some limitations. We did not have access to clinical progression scores or causes of death and, therefore, relied on all-cause mortality risk as an indicator of disease progression. Consequently, some of the results may not reflect disease modification but rather effects on general mortality, which we explored in the general population. Because aging is the strongest risk factor of PD, some pathophysiologic processes may be common to both PD and aging. This overlap may explain the observed associations in both populations, although most showed stronger effect sizes in the PD population. It is possible that drugs that displayed equally strong or a stronger association with mortality in the general population compared with the PD population do not have a PD-specific effect.
Among the identified drugs were macrolides, opioids, and melatonin, all of which have been demonstrated to influence mitochondrial quality-control mechanisms, which are central to the suggested pathologic origins of PD, but also to aging in general
The adjusted mortality curves for the drugs with a more pronounced association with mortality in the PD population compared with the general population are displayed in Figure 2. The adjusted mortality curves for the drugs with similar or larger effect sizes in the general population are displayed in eFigure 3.
Editorial: It Is Time for Drug Repurposing in Parkinson Disease
For almost all the 13 compounds displayed in Figure 2 in the article by Tuominen et al., the eight-year adjusted mortality curves for people with PD and healthy controls diverged and had different slopes, suggesting that these compounds may have a disease-modifying role.
6 Likes
I cannot see the melatonin dosing, but to have a good effect needs more than the normal 0.3mg to say 10mg that is thought to be OK.
1 Like
Niagen Bioscience secures exclusive rights for Parkinson’s therapy 2025
The RCT results will be published soon and they might already know that the results are positive?
1 Like
It’s a Norwegian paper. In Norway, melatonin doses below 1 mg are OTC supplements while doses of 1 mg or more are Rx drugs. The paper looked at “prescription drugs” so at least 1 mg of melatonin. It seems that they don’t have more than 10 mg: Medisin - Felleskatalogen
In practice, almost all prescriptions are for Circadin which is melatonin 2 mg XR: https://www.fhi.no/contentassets/b0802ad9303347b682cf6a8fa701ec91/legemiddelforbruket-i-norge-2019-2023-rapport-2024.pdf
I would not think this is a surprising commercial step. I don’t see any payment for it being mentioned.
Great find! Had to clue levothyroxine (T4 thyroid) would do this well. Along with four opioid derivatives.
A few questions
-Any idea on the glucosamine dosage?
-Do you think chondroitin would be of any benefit? (as I’ve been using a combo supplement of glucosamine and chondroitin, but considering to go to glucosamine only next buy.)
-Is there a list of the other antidepressants they tested?
“Mianserin, the only antidepressant associated with reduced mortality in our study, distinguishes itself from other antidepressants by effectively and selectively elevating norepinephrine levels by both blocking its reuptake and stimulating its release.”
I wonder if other norephinephrine reuptake inhibitors would do well. Or other medications that stimulate its release.
Or if it could also be due to histamine blocking (H1 mostly) for Mianserin, since ranitidine was in the list (histamine-2 blocker). Meclizine did good in the ITP study, and it’s H1 blocker. It might have been the only histamine antagonist medication they have tested IIRC.
Check the Norwegian drug database (link above) and see which doses are sold there.
I have no idea what these drugs are sorry.
“We applied the emulated target trial framework and conducted a high-throughput, hypothesis-free screen of all drugs on the Norwegian market initiated by individuals after the diagnosis of PD.”
1 Like
We had longitudinal studies and MR pointing towards beta-blockers being (causally?) linked to a higher risk of PD. Now we know that beta-blockers users who have PD decline faster: Clinical progression and genetic pathways in body-first and brain-first Parkinson’s disease 2025
1 Like
Thanks for the links above, I missed them earlier.
It appears the glucosamine is 400mg pills. It says to take 1 pill 3 times a day, or all 3 pills at one time.
1200mg total.
It also showed a glucosamine/chondroitin/msm (500mg/400/100) pill.
Interesting post about the beta-blockers.
Edit: Was late at night for me.
The glucosamine pharmaceutical appears to be a 1500mg pill.
Seems interesting the connection between nitric oxide and longevity: Tadalafil, ARB, Statin.
Citrulline malate or AAKG (arginine) might be a good addition for nitric oxide and mitochondria (citric acid cycle).
2 Likes
@John_Hemming’s theory vindicated? Just published: Citric acid disassembles α-synuclein fibrils and reduces their cytotoxicity 2025
![]() Korean paper from an okay but not Tier 1 institution + Short publication and not long paper + Mechanistic in vivo model
Korean paper from an okay but not Tier 1 institution + Short publication and not long paper + Mechanistic in vivo model ![]()
Misfolding and aggregation of α-synuclein are associated with the progression of Synucleinopathies including Parkinson’s disease
Citric acid can disrupt the β-sheet structure of α-synuclein fibrils and inhibit additional fibril formation
The molecular docking simulation shows that citric acid exhibits a strong affinity for β-sheet stacking and adjacent regions
Citric acid reduces the cytotoxicity of α-synuclein fibrils
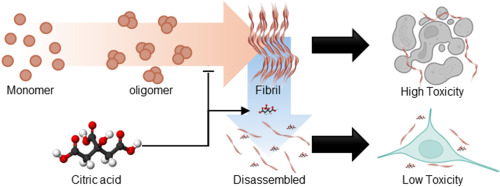
α-synuclein plays a crucial role in regulating neurotransmitter release and synaptic plasticity [1]. However, the misfolding and aggregation of α-synuclein are major hallmarks of Lewy bodies related to progressive neurodegenerative disorders, including Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy [2]. Thus, developing therapeutics that target α-synuclein aggregates is crucial for treating synucleinopathies, as it alleviates neurotoxicity [3]. Herein, we demonstrate that citric acid (citrate, 2-hydroxy-propane-1,2,3-tricarboxylic acid), commonly found in food supplements, can disassemble α-synuclein fibrils and mitigate their neuronal toxicity (Fig. 1A). Our findings reveal that citric acid can effectively disrupt β-sheet structure of α-synuclein fibrils and inhibit further fibril formation. Molecular docking (MD) simulation further supports these results, showing that citric acid exhibits a high affinity for the β-strand and the surrounding region within α-synuclein fibrils. We propose that citric acid holds significant promise for pharmaceutical applications in treating neurodegenerative diseases related to synucleinopathies.
In summary, we demonstrated that citric acid can effectively inhibit the α-synuclein fibril formation and disaggregates existing fibrillar aggregates. It also reduces the cytotoxicity of α-synuclein fibrils and interacts with the β-strand regions and their surrounding residues within the fibril structure. Given that pathological α-synuclein aggregates may originate in the gut, orally administered citric acid could act locally to disassemble them before brain propagation. Notably, although typical plasma citrate levels are around 0.1 mM, gastrointestinal concentrations can transiently reach 10–20 mM after citrus ingestion, supporting the feasibility of our effective dose for gut-targeted intervention.
1 Like
My theory is moreso about acetylation.
This, however, would help particularly with PD
3 Likes
Yes. I find the closing remark about gut concentrations interesting. Worth exploring…
2 Likes
Obviously the gut will be relatively high, but serum is unlikely to get over 0.5mM
1 Like
It’s well known that coffee is associated with a lowest risk of PD. However, RCT failed. A new Mendelian randomization clarifies the situation: Coffee Consumption Is Associated With Later Age-at-Onset of Parkinson’s Disease 2025
Using Mendelian randomization, we identified a significant association between coffee consumption and delayed PD AAO (IVW: OR, 1.91; 95% CI 1.53–2.38; p = 8.072e-09), but no causal association or genetic correlation with PD risk or progression. Our findings suggest a potential causal effect of higher coffee consumption on PD AAO, with no evidence of an association with PD risk or progression.
On another topic and relevant to those taking immunosuppressants such as rapamycin? Prognostic significance of peripheral lymphocyte counts in Parkinson’s disease 2025
PD patients who reached an endpoint had lower lymphocyte counts at baseline.
PD patients with low lymphocyte counts reached an endpoint earlier.
Motor symptoms and lymphocyte count were associated with reaching an endpoint.
They don’t define the threshold between the “low lymphocyte” and “high lymphocyte” groups but they give the groups’ averages (×10^3/μL): 1.10±0.25 (low) vs 1.87±0.44 (high).
Do you have any thoughts on this @John_Hemming?
WBC issues are complicated.
In theory a low aggregate WBC count is pro longevity, but within that a high proportion of lymphocytes is better.
Hence simply looking at the lymphocyte count ignores the conflicting points.
1 Like