One more thing, and I will stop spamming: Mg threonate became popular in the mid 2010s. Magtein was launched in 2012. This Tier 2 Chinese paper found benefits in a rodent model of PD (MPTP) in 2019. 6y later: nothing more. So if dozens of random people are discussing the potential of Mg threonate for PD, surely some researchers are considering it as well. Maybe they’re trying it on a single mouse in their lab, see nothing, give up, and never publish anything. I might be wrong, but I feel like the lack of subsequent publications is a bad sign. The next logical step for the Chinese team that reported a positive result in the MPTP model would be to test it in another rodent PD model (transgenic, α-synuclein, 6-OHDA, mitochondrial dysfunction, etc.) or in another species (C. elegans, zebrafish, etc.) or for another disease (esp. Alzheimer’s). Of course there’s not much money in science to test supplements and other non-patented compounds but researchers want citations, so you have something that seems to work, isn’t it an easy win to try it on another model? Also, Mg Threonate is partially patented via Magtein so the “no one will make money here” argument is weaker. So I’m skeptical…
That’s disappointing. But this is a review, so how much can they screw up - I guess you have to follow all their references to see if they are distorting things; that’s a lot of work just to look at case reports. In general, the benfo excitement has kind of died down a bit. About 16 months ago I heard some rumors that people in Japan have been looking at benfotiamine and PD, but then nothing happened since. But like I said before, I’m very tired with papers that focus on a single molecule. No single molecule is going to “solve” PD, and constantly reading papers with such “findings” just makes me feel hopeless. We need a comprehensive approach starting with fundamentals, otherwise PD will turn into another “cure cancer” dynamic with no end in sight. So, some time ago I decided to just select one area and do a deep dive as far as I possibly can without having a lab. I want to become actually “specialist” (at least by absorbing the literature), in that one part. I selected calcium handling, because it seems to be such a basic part in NDDs in general.
Insofar as single molecules, I try to look at the broader context. For example, I am now convinced that statins are actually a negative for PD. But why? First you have disentangle the effect from the mechanism. The effect is lower LDL (or ApoB) - is that part of the chain? Or is it the mechanism of statins that’s the issue. Focusing on mechanism, as an example, I posted a study about how pitavastatin compared to other statins doesn’t deplete plasma CoQ10. If statins in general deplete CoQ10, is that going to factor into why they’re bad for PD? So it’s not just focusing on a single molecule - as an example of such:
Coenzyme Q10 and Parkinsonian Syndromes: A Systematic Review
So, would that mean that say, pitavastatin - unlike other statins - might not be a PD negative? There’s not enough in the literature to determine much. Exposure in early stages of PD (including before any symptoms), full blown PD, early onset vs late onset.
If you look at supplementing with CoQ10, this doesn’t do anything for PD - but what about supplementing for people who take statins? That has generally been underwhelming too.
So CoQ10 levels are lower in people with PD - but what do we do with that information? That’s the problem with single molecules being measured - because serum CoQ10 levels in PD are normal (or close to), the depletion is in various tissues, lympocytes etc. You don’t know why levels are lower in those tissues - is it downstream of some pathology, is causative itself or merely a marker etc. - that’s why I was interested to see if pitavastatin would exacerbate PD as well, since it doesn’t affect CoQ10 as much, but that’s again a tissue vs plasma thing. If you want to see how crazy these effects are of various statins, look at this (rat) study (free paper):
Influence of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on ubiquinone levels in rat skeletal muscle and heart: relationship to cytotoxicity and inhibitory activity for cholesterol synthesis in human skeletal muscle cells
“This study sought to evaluate and compare the cytotoxicity of statins (cerivastatin, pitavastatin, fluvastatin, simvastatin, atorvastatin and pravastatin) in cultured human skeletal muscle cells (HSkMCs) and the effects on ubiquinone levels in statin-treated rat skeletal muscle and heart. Cerivastatin, the most potent inhibitor of HMG-CoA reductase, showed the strongest cytotoxicity (over 10-fold) among the statins examined, while the effects of the others were in a similar range. In rat experiments, neither pitavastatin nor cerivastatin decreased ubiquinone levels in skeletal muscle, but both dose-dependently lowered ubiquinone levels in the heart. As the rates of reduction by pitavastatin (9.6% at 30 mg/kg) and cerivastatin (9.7% at 0.3 mg/kg) were almost equal, it was estimated that cerivastatin reduced ubiquinone levels in the rat heart approximately 100-fold more strongly than pitavastatin, based on the effective doses. We found that cerivastatin showed the most potent cytotoxicity in HSkMCs and strongly lowered ubiquinone levels in the rat heart.”
So, CoQ10 levels in skeletal muscle tissue (where people complain about myalgia) were not affected by either cerivastatin or pitavastatin, both lowered levels in the heart, but cerivastatin reduced levels 100-fold more strongly than pitavastatin dose effectively?! And cerivastatin was vastly more cytotoxic.
Bottom line, you have to measure the levels of any single molecule in every tissue and you might find that the effects even in the same class of medications (say, statins), to be dramatically different.
Anyhow, it’s nice to come up for air from all these mechanistic studies and into the calm waters of outcome studies, where you just have to battle confounders. And in outcomes, benfotiamine is not shining - of course, maybe in combination with other stuff it might be of help.
It’s mostly LDL. Per MR. Glucose dysregulation might play a role as well but it’s mostly LDL lowering.
@adssx and @CronosTempi I appreciate the ongoing dialogue + findings discussions, as I’m one of those fighting early-onset PD. My current simplifying protocol is diet (Mediterranean/pescetarian/minimal dairy + red meat) and exercise (resistance and 80%+ heart rate cardio).
I take B-12 and fish oil omega-3 supplements too, plus rapa (hence being here!). I guess I’ll be measuring any success at a decadal level of resolution.
Sorry to hear that. You might be interested in my “strategy”: Parkinson's disease - #429 by adssx
I changed a bit my “stack”. It’s now (besides the usual sleep, diet, exercise, stress management, etc.):
Unchanged:
- Vaccinations (esp. shingles and tetanus)
- Dapagliflozin 10 mg
- Telmisartan 80 mg
- Amlodipine 5 mg
- Lithium orotate 1 mg
- Methylcobalamin (because I’m a bit deficient)
Added:
- UDCA 2x500 mg: Good evidence + Ongoing RCT + Subjective benefits felt. I tried TUDCA, and it’s not as good as UDCA.
Testing:
- Sirolimus: 6 mg every two weeks, backed by some evidence and gives me an energy boost
- Lactoferrin (native bovine): this one really boosts me but there’s very little evidence, might be placebo or something else? It did increase my transferrin saturation (TSAT) a lot while not moving much other iron-related biomarkers. See: Lactoferrin: A Milk-Derived "Immunoceutical" Reverses the Clock on Inflammaging
- K2 MK-7: saw some benefits but TBC Menaquinone-7 (MK-7): An interesting molecule to power up the mitochondria
Dropped:
- Ezetimibe: My apoB is ~70 mg/dL, and cholesterol-lowering seems to be a net negative in PD. Choose your poison…
- Selegiline: Lifespan extension not confirmed + potential negative cardiac effects
- Theracurmin: Not enough evidence + no perceived subjective benefits
- Terazosin: I gave it a try, some symptomatic benefits but I’m not convinced by the long-term cardiac
- Tanganil: didn’t feel much, evidence weak
- Semaglutide: felt worse on it, exenatide phase 3 failed
Undecisive:
- Ambroxol
- Melatonin
- Intermittent hypoxia: Oxygen, hypoxia and hyperoxia
Interesting:
- Monepantel: Monepantel: a brain-penetrant mTOR inhibitor
If you take B12 take methylcobalamin.
Omega-3: There’s 0 evidence in PD. Actually, there’s 0 evidence in any condition, with the exception of EPA (eicosapentaenoic acid) in those with high triglycerides.
All good stuff to check in on. Here’s my stack/recent history:
- shingles/tetantus updated in last 2y
- telmisartan 80mg daily
- rapa 8mg / 8d
- liraglutide/tirzepatide 15u/8d
I have elevated homocystiene levels that have dropped slightly since starting peptides, but that seems speciously correlated. Any thoughts on methyl guard?
Evidence for all those vitamins seems very weak to me (in general for longevity and for PD in particular). That’s why I don’t take it. I take methylcobalamin (forgot it in my previous message, just edited it), but only because otherwise I’m borderline deficient.
Liraglutide/tirzepatide: Did you notice any benefit? Why liraglutide? In the phase 3 exenatide trial people on exenatide were slightly worse off than placebo. I think GLP1-RAs might work better on people with some glucose regulation issues. What was your HOMA-IR before starting GLP-1RAs?
I have decided (following test results) to take a cut down supplement stack on fasting days of b(5,6,7,9,12) and d3

A drug–microbiome–drug interaction impacts co-prescribed medications for Parkinson’s disease
https://www.nature.com/articles/s41564-026-02299-2
" Iron binding to COMT-I drugs modulates their antibacterial activity
Previous literature describes the iron chelating ability of catechols34,35, including nitrocatechols such as entacapone30. Similar to previous reports of other nitrocatechols35, incubation of tolcapone with either ferrous iron (Fe2+ in FeSO4; Fig. 2a) or ferric iron (Fe3+ in FeCl3; Fig. 2b) results in a red shift in ultraviolet–visible (UV–vis) light absorbance, consistent with iron binding. This shift does not occur when M1 or M2 is incubated with ferrous or ferric iron, suggesting a lack of iron binding by these metabolites (Extended Data Fig. 3a,b)."
How does this look when you throw in lactoferrin?
Combination Disease-Modifying Therapy for Neurodegenerative Diseases Using Repurposed Drugs 2026
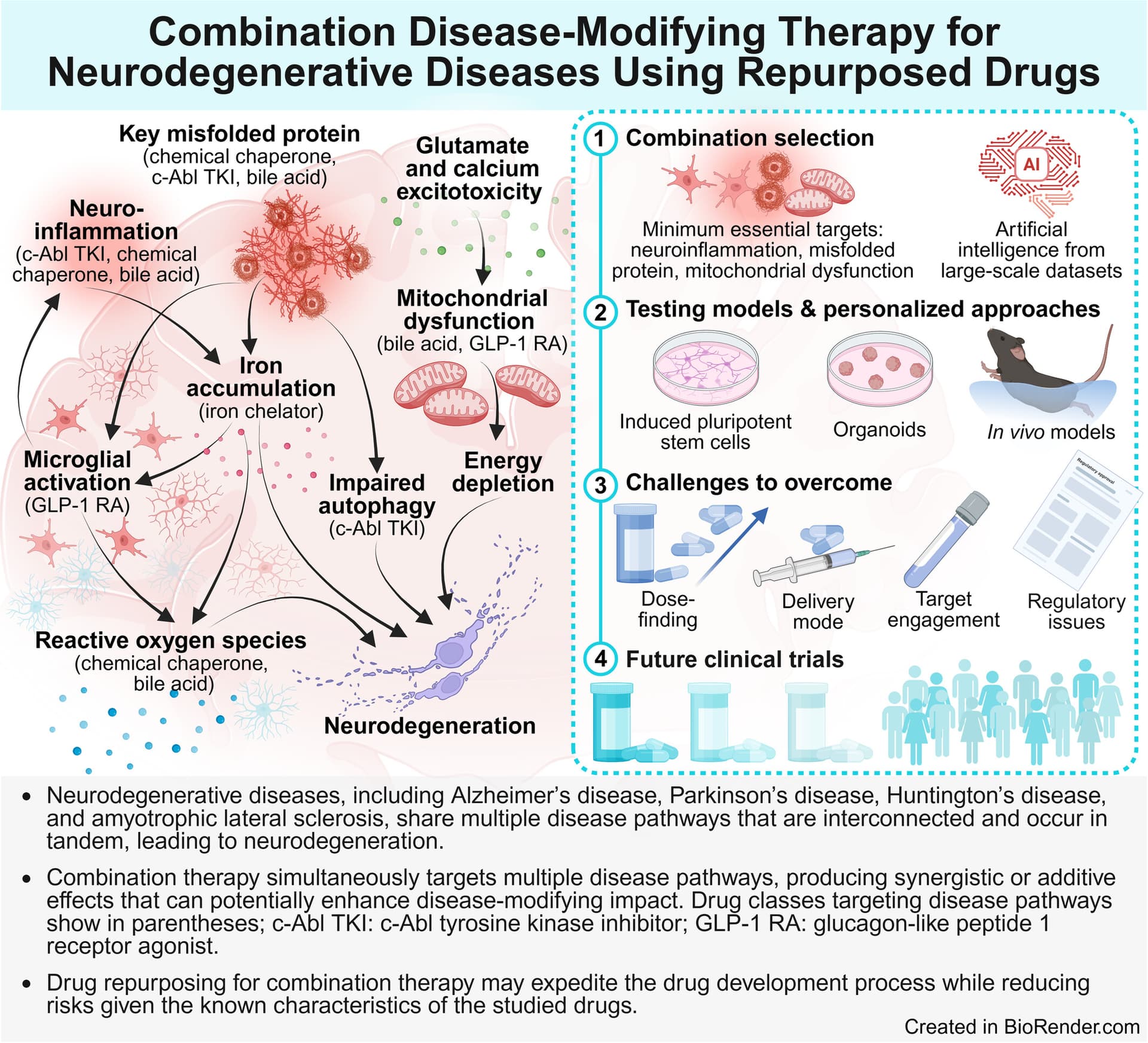
That’s kind of my strategy as well with my combos… Is it working? God knows!
Apologies if this has been posted before…

https://x.com/Neuroscope_mp/status/2041486879929876484?s=20
clinical trial details: ClinicalTrials.gov
Video:
The discussion centers on the potential of smartphone applications in aiding the monitoring, diagnosis, and detection of Parkinson’s disease, particularly focusing on a keyboard typing application. The speaker highlights their involvement with a company that utilizes technology to track changes in typing behavior, which can serve as a sensitive indicator of disease progression and treatment response. This approach aims to complement existing diagnostic methods such as the FDA-approved SPECT-Ioflopane imaging study, commonly known as the virtual DaT scan, which evaluates the integrity of the nigrostriatal dopaminergic system.
https://www.neurologylive.com/view/use-of-smartphone-apps-for-pd-management
Mysterious ‘compound X’ clears toxic Parkinson’s proteins from brain
A drug known only as compound X helped to remove the problematic proteins associated with Parkinson’s disease from the brains of mice, and improved their balance and mobility
full story: Mysterious ‘compound X’ clears toxic Parkinson’s proteins from brain (New Scientist)
So sorry to hear about your Dx, Antoine. Knowing how detailed your research of the literature is (I appreciate these posts in particular), no doubt you’ll leave no stone unturned.
Article implies this is a repurposed compound. Any idea what the compound is? Supposedly it helps improve deep sleep and glymphatic drainage.
ChatGPT’s best guess:
Based on the reported effects, especially increased slow-wave sleep and glymphatic clearance, this is almost certainly a repurposed CNS-active sleep-modulating drug.
Top candidates:
Suvorexant
Lemborexant
Daridorexant
These fit best because orexin antagonists directly enhance deep sleep, which is strongly linked to glymphatic activity and protein clearance.
Secondary possibilities:
Trazodone
Gabapentin
Less likely, but still plausible due to effects on sleep architecture.
Bottom line: an orexin antagonist is the most likely class.
Google’s Gemini suggested Dexmedetomidine (brand name Precedex) is an FDA-approved drug (used in ICU for sedation and for Delirium management) that has been shown in studies to boost slow brain waves and enhance glymphatic drainage, possibly in combination with Midodrine. A March 2026 study found this combination (ACX-02) effectively clears amyloid and tau in humans compared to placebo: Pharmacological enhancement of glymphatic function in humans increases the clearance of Alzheimer’s disease-related proteins