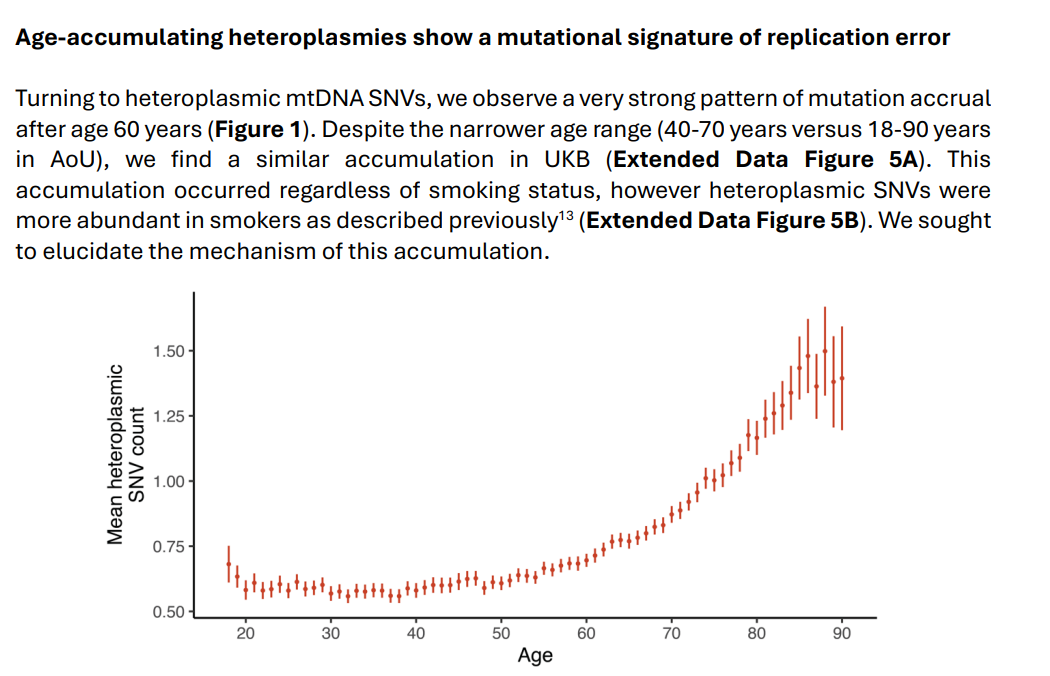
ABSTRACT
One of the strongest signatures of aging is an accumulation of mutant mitochondrial DNA
(mtDNA) heteroplasmy. Here we investigate the mechanism underlying this phenomenon by
calling mtDNA sequence, abundance, and heteroplasmic variation in human blood using
whole genome sequences from ~750,000 individuals. Our analyses reveal a simple, two31 step mechanism: first, individual cells randomly accumulate low levels of “cryptic” mtDNA
32 mutations; then, when a cell clone proliferates, the cryptic mtDNA variants are carried as
33 passenger mutations and become detectable in whole blood. Four lines of evidence support
34 this model: (1) the mutational spectrum of age-accumulating mtDNA variants is consistent
with a well-established model of mtDNA replication errors, (2) these mutations are found
primarily at low levels of heteroplasmy and do not show evidence of positive selection, (3)
high mtDNA mutation burden tends to co-occur in samples harboring somatic driver
mutations for clonal hematopoiesis (CH), and (4) nuclear GWAS reveals that germline
variants predisposing to CH (such as those near TERT, TCL1A, and SMC4) also increase
mtDNA mutation burden. We propose that the high copy number and high mutation rate of
mtDNA make it a particularly sensitive blood-based marker of CH. Importantly, our work
helps to mechanistically unify three prominent signatures of aging: common germline
variants in TERT, clonal hematopoiesis, and observed mtDNA mutation accrual.
Is an interesting paper. There is clearly a balance between mtDNA damage caused by replication errors and that caused by ROS. I would argue that the cells with more ROS have more mtDNA damage from ROS.
Blood cells (one assumes mainly white blood, but also platelets) are going to be different to high energy neurons.
This is an interesting extract.