https://www.biorxiv.org/content/10.64898/2026.06.26.734787v1
Obviously not news to anyone who reads this forum
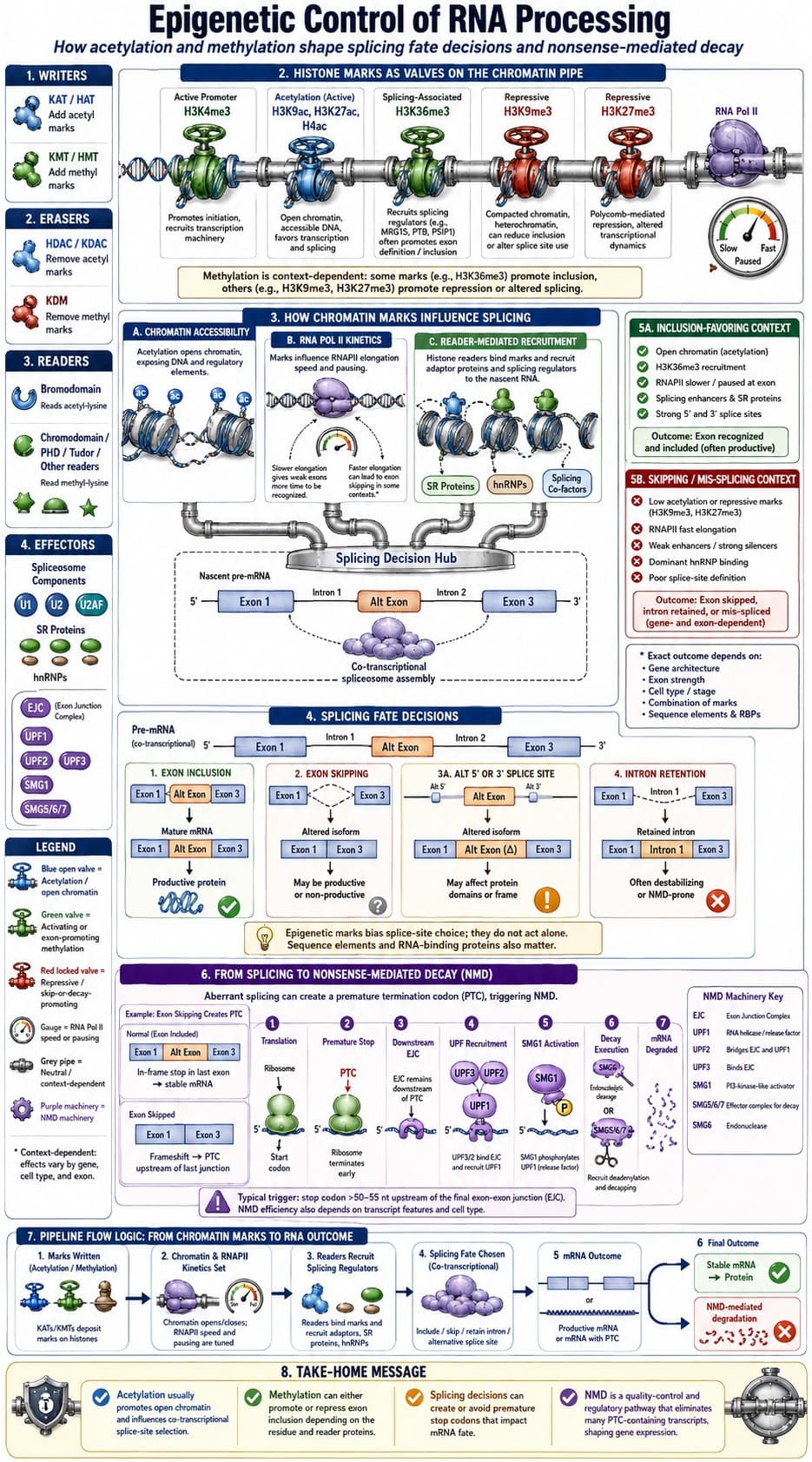
chatGPT(5.5paid):
Summary
The paper argues that mammalian aging is accompanied by a genome-wide decline in the fidelity of alternative splicing, which the authors call “splicing degeneration.” Rather than simply cataloguing age-associated splice changes, they try to distinguish splice changes likely to damage protein function from those that may be neutral or adaptive.
The authors analyse large RNA-seq datasets from humans and mice, especially GTEx human tissues. They calculate PSI values — percent-spliced-in — for alternative splicing events, then compare older samples with younger reference samples. They find that the number of differential splicing events rises with age across many tissues. They also build splicing-based aging clocks, showing that splicing patterns can predict chronological age reasonably well.
A central step is their classification of splicing events as potentially damaging if they:
- introduce a premature termination codon, especially through intron retention;
- cause a frameshift;
- alter a region overlapping a protein domain.
Using this framework, they find that age-associated splicing events are enriched for damaging consequences. These events occur disproportionately in conserved genes and in genes involved in RNA metabolism, RNA splicing, antigen presentation and related processes.
They then define a quantitative splicing degeneration score: the proportion of damaging alternative-splicing events present in a sample. This score increases with age in many human and mouse tissues. Lung, spleen and small intestine appear among tissues with relatively consistent age-associated increases.
The paper also claims that splicing degeneration is modifiable. In reanalysed datasets, rapamycin, everolimus and calorie restriction tend to reduce splicing degeneration. Metformin and S6K1 deletion show weaker or non-significant effects. This is used to argue that splicing degeneration is not merely a passive correlate of chronological time, but may be linked to modifiable aging biology.
Mechanistically, the authors focus on splicing factors. They identify splicing factors whose expression or own splicing changes with age, including members of the SRSF and hnRNP families. They propose a “self-regulation” model: age-related mis-splicing of splicing factors contributes to broader splicing degeneration. They combine GTEx correlations, ENCODE knockdown RNA-seq, and eCLIP binding data to identify candidate regulators, including SRSF1, SRSF5, U2AF1, PTBP1, QKI, PUF60, SF3B1, HNRNPA2B1, HNRNPC, PRPF8 and others.
Finally, they use transcriptomic aging clocks to show that knockdown of many splicing factors increases predicted transcriptomic age. This supports the idea that splicing factor dysfunction can push gene-expression states toward an older profile.
Claimed novelty
The main novelty is not the observation that splicing changes with age; that is already known. The novelty is the attempt to define a functional, genome-wide measure of age-related splice damage.
The paper’s key novel contributions are:
1. “Splicing degeneration” as a formal aging phenotype
The authors move from “alternative splicing changes with age” to “damaging isoforms accumulate with age.” That is a stronger conceptual claim.
2. A quantitative degeneration score
They propose a score based on damaging splice outcomes: premature stops, frameshifts and domain-disrupting events. This makes splicing degeneration measurable across datasets, tissues and interventions.
3. Cross-species and multi-tissue integration
They analyse human GTEx, mouse datasets, Tabula Muris, cancer datasets and intervention datasets. This breadth strengthens the case that the phenomenon is general rather than tissue-specific.
4. Link to longevity interventions
The claim that rapamycin, everolimus and calorie restriction reduce splicing degeneration is particularly interesting, because it links splice fidelity to interventions already known to affect aging pathways.
5. Splicing factors as both victims and drivers
The “self-regulation” model is useful: splicing factors themselves become mis-spliced or mis-expressed with age, which may amplify downstream splicing disorder.
6. Connection to transcriptomic age
Showing that splicing-factor knockdowns increase transcriptomic age connects splice regulation to broader aging signatures rather than treating it as an isolated molecular defect.
Critique
The paper is important and highly relevant to aging biology, but several claims need caution.
First, “damaging” is computationally inferred, not experimentally proven. A frameshift, premature stop or domain overlap is plausibly damaging, but not all such events will produce stable protein products. Many may be degraded by nonsense-mediated decay or may occur at low abundance. Conversely, some splice changes classified as damaging could be regulated, adaptive or tissue-specific.
Second, bulk RNA-seq is vulnerable to cell-composition effects. Aging tissues often change in immune infiltration, fibrosis, senescent-cell burden and cell-type proportions. Some apparent splicing degeneration may reflect a different mixture of cells rather than splice failure within the same cells. The authors acknowledge this, but it remains a major limitation.
Third, GTEx is cross-sectional and post-mortem. GTEx samples differ in cause of death, ischemic time, tissue quality, RNA integrity and donor health. The model adjusts for some variables, but residual confounding is likely. Age-associated splice changes in GTEx may partly reflect illness, agonal state or sample handling.
Fourth, the PSI thresholds are somewhat arbitrary. The score uses PSI > 0.3 for damaging intron retention and PSI < 0.7 for damaging exon/skipping-related events. The authors test cutoffs, but the biological meaning of these thresholds is not obvious. A small change in a highly expressed essential gene may matter more than a large change in a low-expression gene.
Fifth, protein-level validation is limited or absent. The argument is about functional decline of protein products, but most evidence is transcriptomic. Ribosome profiling, proteomics, long-read RNA-seq, isoform-specific protein detection, or functional rescue experiments would make the case much stronger.
Sixth, the intervention analysis is suggestive but not definitive. Rapamycin-associated reductions in splicing degeneration are interesting, but these are reanalyses of heterogeneous datasets. Differences in cell type, dose, duration, sequencing method and baseline state could influence the result. It does not prove that rapamycin extends lifespan by restoring splicing fidelity.
Seventh, Mendelian randomisation is a bold addition, but interpretation should be cautious. Splicing QTLs can have pleiotropic effects on expression, chromatin or nearby genes. Inferring that particular splicing events causally influence Horvath age or lifespan is plausible but not settled.
Eighth, the “splicing degeneration” label may be partly value-loaded. Some age-associated splice changes may be adaptive stress responses, tumour-suppressive shifts, immune remodeling, or compensatory changes. The term “degeneration” is appropriate for the damaging subset, but perhaps too broad if applied to all age-related AS change.
Overall assessment
This is a strong and conceptually useful paper. Its main value is that it reframes age-related alternative splicing as a measurable form of molecular damage, rather than just another transcriptomic correlate of age. The link with rapamycin and calorie restriction is especially interesting.
The paper is best viewed as a large-scale computational hypothesis-generating study. It makes a persuasive case that splice fidelity declines with age and may be modifiable, but it does not yet prove that splicing degeneration is a primary driver of aging or functional decline.
The next decisive experiments would be:
- single-cell or sorted-cell validation to separate cell-composition effects from within-cell splice decline;
- long-read RNA-seq to confirm full-length isoform changes;
- proteomic or ribosome-profiling evidence that damaging isoforms alter protein output;
- intervention experiments where specific splicing factors are restored in aged tissues;
- tests of whether correcting selected age-damaging splice events improves cellular or organismal function.
For your broader acetylation/splicing hypothesis, the paper is quite supportive: it strengthens the idea that aging involves a genome-wide deterioration in isoform control, especially affecting conserved and functionally important genes. It does not directly prove an acetyl-CoA or histone-acetylation mechanism, but it fits well with a model in which age-related transcriptional and chromatin changes impair co-transcriptional splicing fidelity.