As probably everyone here is already aware, AKG is a promising supplement that produces lifespan increase in middle-aged female mice, healthspan increase in middle-aged female and male mice, and has ongoing ITP support (20’ and 22’ cohorts).
There’s also some human data, where 7 months of an AKG-based formulation led to an average 8 year reduction in TruAge DNA methylation assay. Then there’s the ABLE trial, which I believe is ongoing.
Despite quite a few people on this site taking AKG, there hasn’t been much discussion of it’s role as a co-substrate of the TET enzymes. I wanted to call more attention to this role, as the TET enzymes may play a crucial role in longevity.
Above paper found that TET1 functions primarily at the shores of promoter-associated CpG islands. Approximately 70% of vertebrate gene promoters contain CpG islands, and as their hypermethylation can lead to stable gene silencing, it’s imperative to prevent methylation from spreading past the shores and onto the CpG island.
The red trace in figure below reflects this function, where the shores (i.e the first couple kb outside of the CpG island) contain peaks of 5hmC.
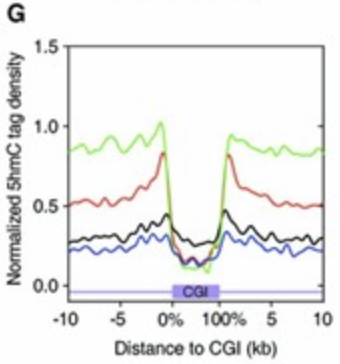
Additionally, relative to catalytically dead TET1 controls, TET1 overexpression doesn’t change the pattern of DNA methylation, nor does it change global methylation levels, nor does the increased demethylation significantly alter transcription.
The upshot of this is that increased TET1 activity (via overexpression and/or increased co-substrate concentration) might function as an epigenetic guardian of many gene promoters.
One potential issue with promiscuous demethylation (as seen with TET1 mutants lacking the CXXC domain needed to bind unmethylated CpG islands) is DNA repair stress. This is because TET1 demethylation requires the base excision repair (BER) pathway for active reconversion to unmodified cytosine. Excessive levels of these BER intermediates (as occurs with overexpression of TET1 mutant) corresponds to increased numbers of abasic sites and SSBs. These lesions activate the DNA damage response (during which time the rate of epigenetic aging might be increased), while the SSBs lead to DSBs in replicating cells.
In light of this, we might want to enhance efficiency of BER pathway if we’re aiming to increase TET1 activity. This might be accomplished via SIRT6 activation, which increases BER efficiency in a PARP1-dependent manner.
Of note, there is a complex interplay between TET1 and PARP1. PARP1 increases TET1 transcription, and also stimulates TET1 activity via PARylation. On the other hand, TET1 activity is inhibited by its noncovalent interaction with PARs. Additionally, TET1 may stimulate PARP1 activity.
In vivo, it seems that the PARP1’s positive regulation of TET1 activity outweighs the negative regulation, since PAR inhibitors lead to decreased 5hmC levels.
So perhaps a cocktail to test would be AKG (increased TET1 activity), SIRT6 activator (increased PARP1 activity), and NAD booster (NAD is a co-substrate for both SIRT6 and PARP1-mediated ADP-ribosyltransferase reactions).
Humans also might stand to benefit from this more than mice, see for example
Our results show that human liver had higher overall expression of genes encoding proteins involved in the BER, HRR, NHEJ, and MMR pathways; however, only the BER pathway expression was significantly higher (p=0.0010). [ref]
Further supporting this, PARP1 assay in mononuclear blood cells (i.e T cells, B cells, NK cells, and monocytes) using radiolabeled NAD shows strong correlation (r=0.84) between PARP1 activity and max lifespan across 13 mammalian species. [ref] Partially this could be due to enhanced BER efficiency, but additional mechanisms (e.g enhanced DSB repair) are likely at play.
A few practical questions:
-
Would arginine-AKG be a superior choice, to avoid excessive calcium supplementation?
-
Would lower doses of AKG (<10,000 ppm) be superior in mice? The lower expression of BER pathway genes in mice liver and low PARP1 activity in rat mononuclear blood cells suggests a reduced ability of mice to prevent a DNA damage response in response to drastic increases in TET1 activity.
-
Would extended release or taking multiple smaller doses throughout the day be preferable? From this perspective, it certainly seems so. Probably this would reduce the risk of promiscuous demethylation and the associated DNA damage response, while also maintaining a more constant increase in TET1 activity throughout the day.
I’ll expand on this post as time allows, as this is already becoming quite a wall of text.
