Is Ferroptosis the Mechanistic Bridge Connecting Iron Dysregulation to Muscle Wasting and Functional Decline in Aging?
Paper here: https://onlinelibrary.wiley.com/doi/10.1111/acel.70367
First published: 14 January 2026
Institution: Department of Physiology and Aging, University of Florida, USA; Università Cattolica del Sacro Cuore, Italy Journal: Aging Cell
This review article advances a “Ferroptosis Bridge” hypothesis, proposing that age-related muscle wasting (sarcopenia) is not merely a result of wear and tear, but a specific form of iron-dependent cell death known as ferroptosis. The authors argue that as we age, skeletal muscle—which naturally holds 10-15% of the body’s iron—loses its ability to regulate iron homeostasis. This leads to an accumulation of labile iron (free Fe2+), which interacts with hydrogen peroxide via the Fenton reaction to generate hydroxyl radicals. These radicals aggressively attack polyunsaturated fatty acids (PUFAs) in cell membranes (lipid peroxidation), causing membrane rupture and cell death.
Crucially, the paper distinguishes this process from general oxidative stress or apoptosis. It posits that the convergence of three aging hallmarks—iron dyshomeostasis, mitochondrial dysfunction, and impaired antioxidant defenses (specifically GPX4 and GSH)—creates a “pro-ferroptotic milieu”. This environment compromises muscle energetics and regenerative capacity. The authors suggest that targeting this specific pathway via iron chelation or lipid peroxidation inhibitors could offer a more precise intervention for sarcopenia than generic antioxidants, which have largely failed in clinical trials.
Impact Evaluation The impact score of this journal is ~7.8 (JIF), evaluated against a typical high-end range of 0–30+(where 30+ is elite general science like Nature), therefore this is a High impact journal within the specialized field of Geriatrics and Gerontology.
2. Mechanistic Deep Dive
The authors construct a specific “lethality pathway” for aging muscle that moves beyond generic “wear and tear.”
-
The Ferroptosis Trigger (Iron + Lipids): The core pathology is the intersection of Labile Iron Pool (LIP) expansion and PUFA-containing phospholipids. Aging downregulates Ferroportin (iron exporter), trapping iron inside the myocyte.
-
The Failed Guardian (GPX4): Under healthy conditions, Glutathione Peroxidase 4 (GPX4) utilizes Glutathione (GSH) to neutralize lipid hydroperoxides. In aging muscle, GPX4 expression declines, removing the “brake” on ferroptosis.
-
The Senescence Loop (Hepcidin Axis): The paper identifies a systemic loop where “Inflammaging” (elevated IL-6) triggers the liver to produce Hepcidin. Hepcidin degrades Ferroportin on muscle cells, forcing iron retention. This creates a feed-forward loop: Senescent cells secrete cytokines → Hepcidin → Iron Retention → Ferroptosis →DAMP release → More Inflammation.
-
Organelle Crosstalk (Ferritinophagy): The review highlights NCOA4-mediated ferritinophagy (autophagic degradation of ferritin) as a double-edged sword. While necessary for iron recycling, dysregulated ferritinophagy in aging releases excessive free iron, overwhelming mitochondrial buffers and triggering the Fenton reaction.
3. Therapeutic Interventions & Biohacks
The review categorizes interventions into three tiers based on the mechanism:
-
Tier 1: Iron Chelation (Direct):
- Agents: Deferoxamine, Deferiprone.
- Mechanism: Binds excess labile iron, preventing the Fenton reaction.
- Efficacy: Reduced muscle loss in sarcopenic mice.
-
Tier 2: Lipid Peroxidation Inhibitors (Specific):
- Agents: Ferrostatin-1, Liproxstatin-1, Vitamin E (limited), Vitamin K (potential).
- Mechanism: Acts as radical-trapping antioxidants (RTAs) specifically within lipid membranes.
- Note: Generic antioxidants (Vit C) fail because they do not specifically target lipid radicals.
-
Tier 3: Lifestyle (Upstream):
- Action: Resistance training and Nordic walking.
- Mechanism: Reduces serum ferritin and enhances antioxidant defense (GPX4/Nrf2).
4. Novelty
-
Specific Definition of Death: It reframes sarcopenia not as “atrophy” (shrinkage) but as “regulated necrosis” (ferroptosis), distinct from apoptosis (which is rare in aged muscle) and necroptosis.
-
The “Double-Hit” Theory: Proposes that muscle wasting requires both iron accumulation and proteostatic collapse (ER stress), identifying why simple iron supplementation or simple antioxidants fail in isolation.
5. Critical Limitations
-
Biomarker Vacuum: The authors admit a critical “Hard Fail” in current diagnostics: there are no specific biomarkers for ferroptosis in humans. Markers like Ferritin and 4-HNE are non-specific indicators of oxidative stress. Without a unique signature (like specific oxidized phospholipids), clinical diagnosis is currently impossible.
-
Translational Gap: The majority of mechanistic proof comes from rodent models or cell cultures where ferroptosis is chemically induced (e.g., by erasing GPX4). Natural aging in humans is messier; the “clean” ferroptosis signal seen in mice may be drowned out by comorbidities in humans.
-
Therapeutic Toxicity: Iron chelators have narrow therapeutic windows. Over-chelation risks anemia and mitochondrial failure (mitochondria need iron for ATP). The review correctly notes that using thalassemia drugs for sarcopenia is high-risk.
-
Data Quality: As a review, this paper generates no new data. It relies on the validity of cited studies, some of which use “ferroptosis” loosely to describe general lipid peroxidation.
Biohacker Verdict: This is a high-confidence hypothesis with medium-confidence evidence. The pathway is biologically sound, but the tools to measure and treat it safely in humans are not yet mature.
-
Hypothesis Confidence: High
-
Translational Readiness: Low
Part 4: Actionable Intelligence
The Translational Protocol
-
Primary Candidate: Deferiprone (DFP)
-
Rationale: It is the only oral iron chelator with blood-brain barrier permeability and clinical precedent for neurodegeneration (PKAN) that is also cited for sarcopenia in mice.
-
Human Equivalent Dose (HED) Calculation:
-
Source Data: The key mouse study (Bose et al., 2025, J Cachexia Sarcopenia Muscle) used 25 mg/kg/day in Klotho mice to prevent sarcopenia.
- The Math:
HED(mg/kg)=Animal Dose(mg/kg)×(Human KmAnimal Km)
HED=25×(373)≈2.03 mg/kg
-
Adult Human Dose (75 kg person): 2.03 mg/kg×75 kg≈152 mg/day.
-
Standard Clinical Dose: For iron overload/Thalassemia, the standard dose is 75 mg/kg/day (approx. 5,600 mg/day).
-
Biohacker Insight: The “anti-aging” HED (152 mg/day) is roughly 3% of the standard clinical dose. This suggests a “micro-dosing” strategy might be safer and sufficient for maintenance, though unproven in humans.
-
Pharmacokinetics (PK/PD):
-
Bioavailability: High (~78% urinary recovery). Rapid absorption.
-
Half-life: Short (t1/2≈1.5−2 hours).
-
Implication: Requires divided doses (b.i.d or t.i.d) to maintain chelation, though the “micro-dose” strategy might aim for pulsed “iron sweeping” rather than constant suppression.
-
Safety & Toxicity Profile:
-
Black Box Warning: Agranulocytosis (sudden drop in white blood cells) occurs in ~1-2% of patients. Neutropenia in ~5%.
-
Monitoring Protocol: FDA mandates weekly Absolute Neutrophil Count (ANC) monitoring.
-
Liver/Kidney: Gastrointestinal distress is common. Zinc deficiency can occur (chelates Zinc too).
Biomarker Verification
-
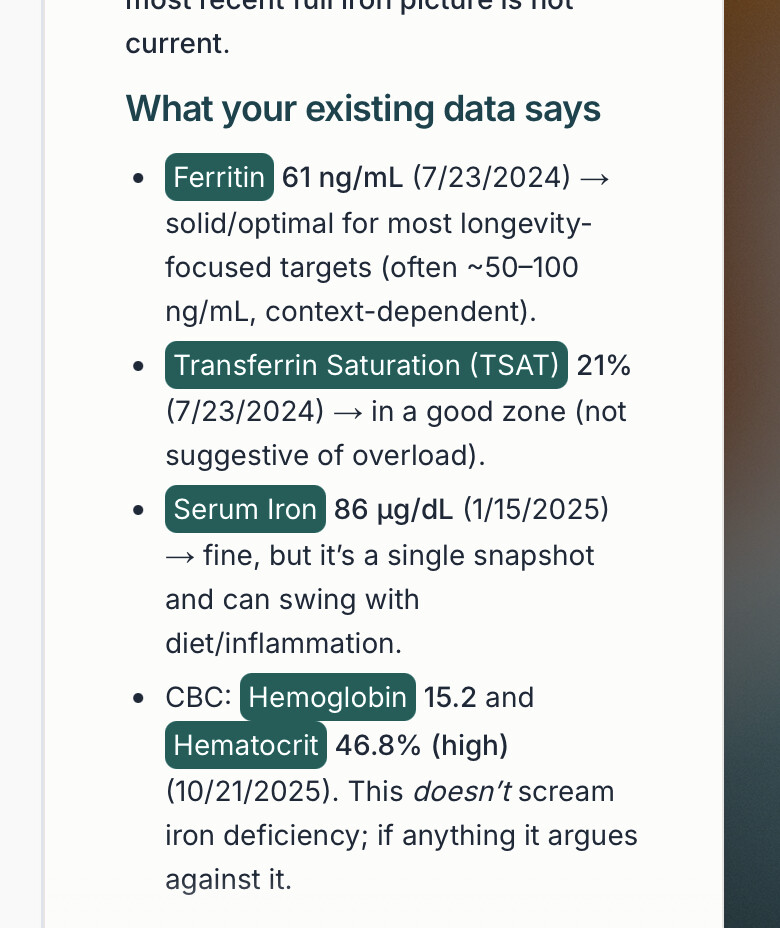
Primary Target Engagement: Serum Ferritin (Goal: Reduction, but not below <30 ng/mL).
-
Downstream Verification:
-
Transferrin Saturation (TSAT): Should drop but stay >20%.
-
Lipid Peroxidation Markers: Oxidized LDL (OxLDL) and F2-Isoprostanes (urine test) are the best available clinical proxies for ferroptosis reduction.
-
Functional: Grip strength (Dynapenia) is the clinical endpoint for sarcopenia.
Feasibility & ROI
-
Sourcing:
-
Status: Rx Only (Ferriprox).
-
Research Chemical: Available but high risk due to purity needs for chronic use.
-
Supplements: None match this mechanism (IP6 is a weak chelator; Curcumin is a weak iron chelator).
-
Cost:
-
Retail: ~$4,000 - $6,000/month for standard clinical dose (Thalassemia).
-
Micro-dose Estimate: If effective at 3% dose, cost theoretically drops to ~$150/month (if compounding/splitting were possible/legal), but shelf-availability is limited to 500mg/1000mg tabs.
Part 5: The Strategic FAQ
1. “Does the ‘Ferroptosis Bridge’ theory hold up if I don’t have iron overload (e.g., normal ferritin)?” Answer: Unclear. The paper argues that labile iron (free floating) is the culprit, not just total storage iron (Ferritin). You can have “normal” ferritin but high labile iron if your antioxidant defense (GPX4) is weak. However, chelating someone with normal/low iron carries a high risk of anemia.
2. “Can I just take Vitamin E to stop lipid peroxidation instead of risky chelators?” Answer: Likely No. Clinical trials of Vitamin E for sarcopenia are largely disappointments. Standard Vitamin E doesn’t penetrate lipid membranes efficiently enough to stop the “ferroptotic wave.” Novel “Radical Trapping Antioxidants” (RTAs) like Liproxstatin-1 are needed but are not human-approved.
3. “If I take Deferiprone, will it strip my Zinc and Copper too?” Answer: Yes. Deferiprone is not perfectly selective. It has an affinity for Zinc (Zn2+). Long-term use requires monitoring of plasma Zinc and potentially supplementation (spaced 4 hours apart).
4. “Is there a ‘Natural’ biohack to mimic Deferiprone without the agranulocytosis risk?” Answer: Pectasol (Modified Citrus Pectin)? Maybe. While not a direct iron chelator, it inhibits Galectin-3 (driver of fibrosis/inflammation). Quercetin and Curcumin have mild iron-chelating properties and are safer, though their potency is orders of magnitude lower than Deferiprone.
5. “How does Rapamycin interact with this pathway?” Answer: Synergistic. Rapamycin inhibits mTOR. High mTOR drives iron uptake (via Transferrin Receptor). By lowering mTOR, you theoretically lower iron influx, complementing the chelator’s removal of existing iron.
6. “Does Resistance Training actually ‘detox’ iron from muscle?” Answer: Yes, indirectly. Exercise upregulates antioxidant enzymes (GPX4, SOD) and improves mitochondrial turnover (mitophagy), effectively “cleaning up” the damage caused by iron, even if it doesn’t directly remove the metal atoms.
7. “I’m on TRT (Testosterone). Does this increase my ferroptosis risk?” Answer: Potentially. Testosterone stimulates erythropoiesis (red blood cell production) and can suppress Hepcidin, potentially increasing iron absorption. Monitoring Hematocrit and Ferritin is crucial for TRT users to avoid “over-juicing” the iron system.
8. “What is the specific ‘Red Flag’ value for Ferritin where I should worry about Sarcopenia?” Answer: Context-dependent. While >300 ng/mL is clinical overload, “optimum” for longevity is debated (often cited as 40-100 ng/mL). High Ferritin + High HS-CRP (inflammation) is the specific “Toxic Combination” linked to frailty.
9. “Can I measure ‘Labile Iron’ directly?” Answer: No. There is no commercial blood test for Labile Plasma Iron (LPI) or Labile Cellular Iron (LCI). We are flying blind using proxies like Ferritin and Transferrin Saturation.
10. “Is this just ‘Anemia of Chronic Disease’ rebranded?” Answer: Nuanced. Anemia of Chronic Disease locks iron away (high Ferritin, low serum iron). This paper argues that inside the muscle cell, that locked-away iron is leaking and causing rust. So, you can be “anemic” in your blood but “iron-toxic” in your muscles.