This paper, and it’s outrageous title, is a troll/marketing piece. Trying to extrapolate a singular mitochondrial enzyme pathway to the “holy grail of human aging”, implicating pretty much every mortality pathway.
The primary author is massively conflicted, even though he declares no conflict. He’s the primary author on many ALCAT1 papers.
He’s the principal of Perenna Pharmaceuticals.
https://www.dnb.com/business-directory/company-profiles.perenna_pharmaceuticals_inc.5a267dfe9ffd5194eb77fa45ef8c3cb7.html
And has a patent application (March 2017 filing) on an ALACT1 inhibitor, assigned to…Perenna Pharmaceuticals. Tell me how that’s not conflicted?
Hmmm…how to get my molecule on the radar?
https://appft.uspto.gov/netacgi/nph-Parser?Sect1=PTO2&Sect2=HITOFF&p=1&u=%2Fnetahtml%2FPTO%2Fsearch-adv.html&r=1&f=G&l=50&d=PG01&S1=20200109136&OS=20200109136&RS=20200109136
He writes in the conclusion: “Although the mitochondrial free radical aging theory was proposed by Dr. Harman more than 65 years ago, it has become the Holy Grail of aging research.”
The oxidative theory of aging is dead.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2789432/pdf/nihms-123940.pdf (2009)
“Over the past eight years, our laboratory (same lab as current author, btw) has conducted an exhaustive study on the effect of under or over-expressing a large number and wide variety of genes coding for antioxidant enzymes. These data demonstrate that almost all alterations in the antioxidant system of mice have no effect on lifespan.In this review, we present the survival data from these studies together. Because only one (the deletion of the Sod1 gene) of the 18 genetic manipulations we studied had an effect on lifespan, our data calls into serious question the hypothesis that alterations in oxidative damage/stress play a role in the longevity of mice.”
I see no clinical trials specific to ALCAT1 pathway drug, yet this acyltransferase was discovered by this lab in 2004.
On ALCAT1 and cancer, the paper is largely irrelevant. I can take care of T2D and CVD thank you, but where is the deferral of cancer? Cancer is most definitely an aging related disease.
“In contrast to its projected role in metabolic diseases, little is known about the regulatory function of CL in the development of cancer. Although the dysregulation of CL metabolism has been observed in several types of cancer, much of the evidence remains correlative and ambiguous. Little is known about the potential role of ALCAT1 in promoting angiogenesis in cancer, which remains to be investigated in future studies”
Holy grail and not delay cancer?
On human neurological “In contrast to insulin-sensitive tissues, such as skeletal muscle
and the heart, TLCL is not the predominant form of CL species in the brain, representingless than 5% of the total CL”. The related passages are ALL references to transgenic mouse models, no human translation.
Another paper on transgenic knockout (ALCAT1-/-) mice fed a high fat diet, modelling obesity:
Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity
https://www.cell.com/action/showPdf?pii=S1550-4131(10)00235-4
“Here, we show that ALCAT1, a lyso-CL acyltransferase upregulated by oxidative stress and diet-induced obesity (DIO), catalyzes the synthesis of CL species that are highly sensitive to oxidative damage, leading to mitochondrial dysfunction, ROS production, and insulin resistance.”
Insulin resistance is not a “mitochondrial causative disease”…it’s a human lifestyle induced metabolic syndrome dysfunction. We’re not trying to “cure a disease” (Type 2 Diabetes) but ameliorate metabolic carnage. So ameliorating the symptoms of T2D via ALCAT1 inhibition, like dozens of other drugs in the market, is a holy grail of aging?
The findings in the ALCAT1/obesity paper ONLY APPLIED TO MALE MICE. "Female mice were unaffected (data not shown)." And ZERO discussion as to why no impact on female mice? Exactly how does a holy grail of aging pathway only apply to males? Fail. Misogynistic chalice!
Unlike Rapamycin which universally extends lifespan in male/female mice!
In another paper, same author:
Pharmacological inhibition of ALCAT1 mitigates amyotrophic lateral sclerosis by attenuating SOD1 protein aggregation (2022)
“Mutations in copper-zinc superoxide dis- mutase (SOD1) are the first identified genetic defect that causes familial ALS [7]. ALS-associated mutations in the SOD1 gene cause SOD1 protein misfolding and aggregation, leading to motor neurons degeneration and death”
So again, a transgenic mouse model of familial ALS.
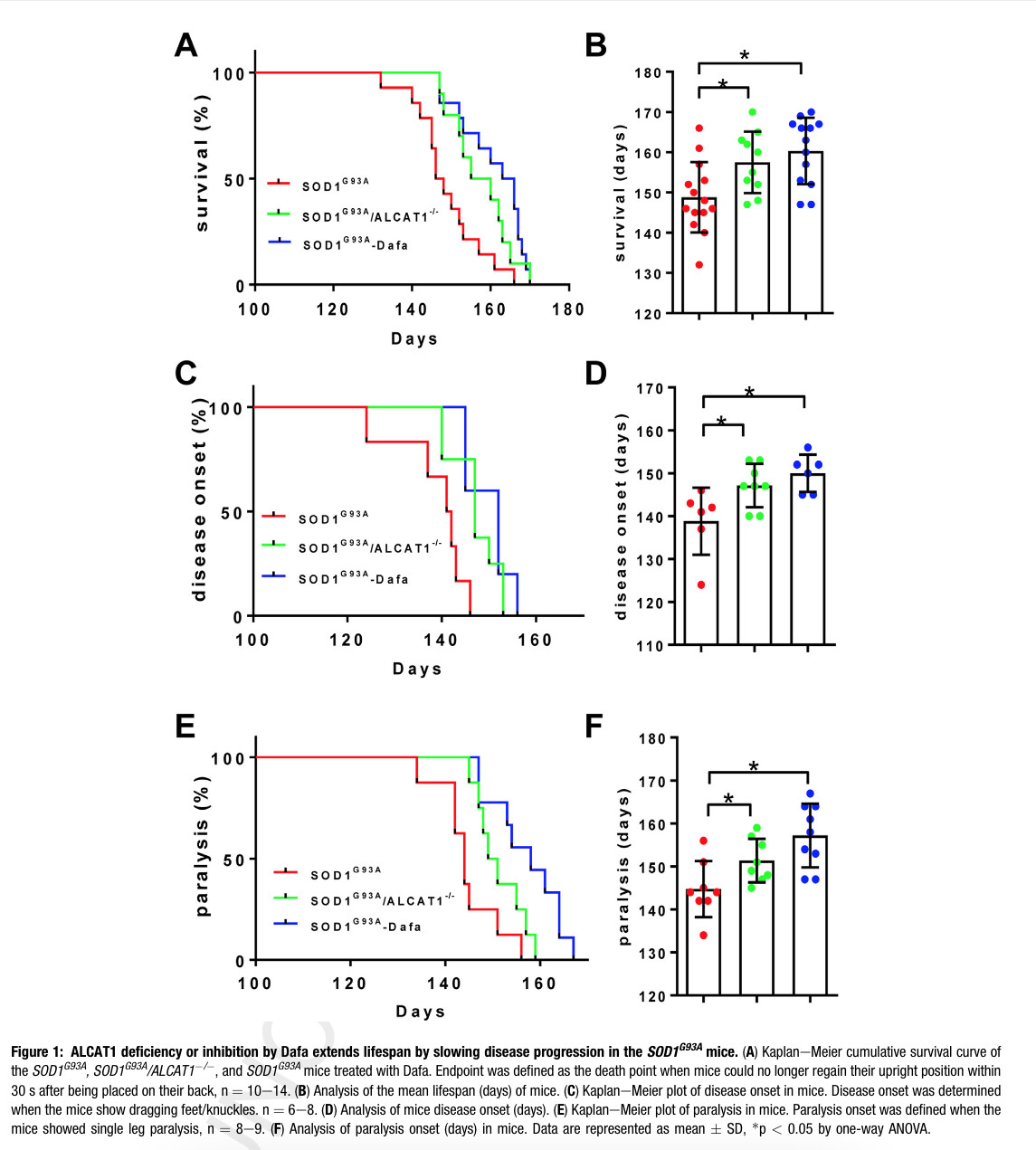
Main findings in image below, showing longevity and disease onset for transgenic ALCAT1 double ablated mice or ALCAT1 inhibition. Less than 10% impact. This is the holy grail?
Here’s another paper on a Parkinsons and ALCAT1. (2019)
“In contrast, ablation of the ALCAT1 gene or pharmacological inhibition of ALCAT1 prevented MPTP induced neurotoxicity, apoptosis, and motor deficits. ALCAT1 deficiency also mitigated mitochondrial dysfunction by modulating DRP1 translocation to the mitochondria. Moreover, pharmacological inhibition of ALCAT1 significantly improved mitophagy by promoting the recruitment of Parkin to dysfunctional mitochondria. Together, these findings identify ALCAT1 as a novel drug target for the treatment of PD”
The “pharmacological inhibition” was via an undisclosed drug A320. In this paper, he full on discloses his conflict:
CONFLICT OF INTEREST
Y.S. is a shareholder of Perenna Pharmaceuticals Inc, a privately held
company which provided the ALCAT1 inhibitor A320 used in this
study
So I went looking for the holy grail…has this group done a study on clean ablated ALCAT1 KO mice, and looked at LONGEVITY. Now that would support the holy grail.
Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression (2012)
https://www.pnas.org/doi/epdf/10.1073/pnas.1120043109
“Using ALCAT1 knockout (KO) mice recently generated in our laboratory (2), we analyzed the effect of ALCAT1 de-ficiency on mitochondrial morphology, mtDNA mass, and mtDNA mutation rate in isolated mouse embryonicfibroblasts(MEFs) and skeletal muscles by EM analysis.”
Uh…where’s the longevity data in aging mice? Non existent.
To summarize the “holy grail paper”, disingenuous tease, shame on the publisher, double shame author/self promoter for not disclosing conflict.