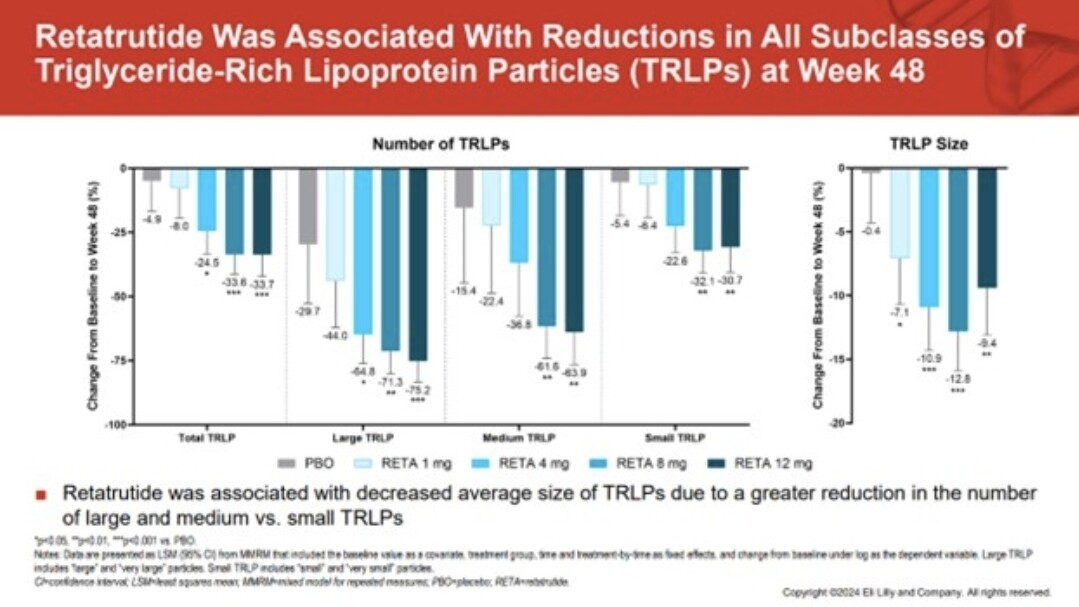
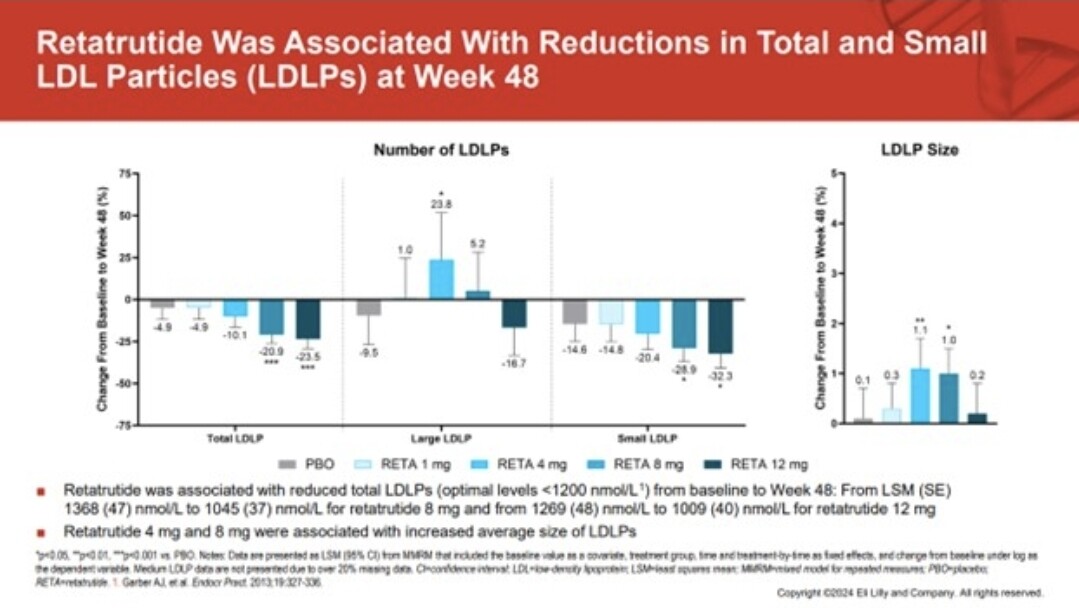
Possibly, but is something else at work for the réduction, and that something seems to be glucagon. Here is the paper where that a slide probably came from
ANGPTL3/8 reductions were observed with 8 and 12 mg retatrutide doses in participants with type 2 diabetes, and with 1, 4, 8 and 12 mg retatrutide doses in participants with obesity or overweight but without diabetes. In both cases, ANGPTL3/8 decreases paralleled retatrutide-induced reductions in TG and LDL-C. In primary human hepatocytes, both glucagon and retatrutide decreased ANGPTL3/8 secretion, and these reductions were blocked with the GCGR antagonist antibody.
Saturated fat usually enlarges LDL particles (shifting from small-dense to large-buoyant) when it replaces carbohydrate, but the effect flips or blunts if you:
Over-feed it to very high levels (> 15 %E) or
Start out with a “pattern B” (sd-LDL–rich) profile and keep triglycerides high.
So saturated fat is not the primary driver of small-dense LDL; hepatic triglyceride output and refined-carb load matter much more.
1 What we mean by “particle size”
LDL subclass
Diameter (nm)
Atherogenicity
Key driver of formation
Large / buoyant (I–II)
26–28
Lower
Direct secretion of cholesterol-rich LDL
Medium (III)
25–26
Intermediate
Hepatic lipase action on TG-rich LDL
Small-dense (IVa–b)
23–25
Highest
High plasma TG → cholesteryl-ester transfer → lipolysis
Key pattern: Saturated fat mainly raises LDL-C by boosting large, cholesterol-rich particles; small-dense particles fall or stay flat unless triglycerides are simultaneously elevated.
3 Why the response flips in some settings
Triglyceride load
• High-carb or over-feeding → ↑ VLDL-TG → more sd-LDL after hepatic lipase trimming.
• Replacing carb with SFA often lowers TG, so fewer sd-LDL precursors.
Baseline phenotype
Pattern B livers already hyper-secrete TG-rich VLDL; flooding them with extra SFA may push both large and small LDL up (Chiu 2017).
Chain length & food matrix Palmitic (C16:0) raises LDL-C most and may nudge sd-LDL in insulin-resistant states; stearic (C18:0) is largely neutral; odd-chain SFAs (C15:0, C17:0) from dairy correlate with larger LDL diameter. (blog.insidetracker.com)
Dose ceiling
Beyond ~15 %E SFA, LDL-receptor down-regulation outweighs the TG-lowering benefit, so total LDL-P climbs in all subclasses.
4 Mechanistic snapshot
SFA ↑ hepatic cholesterol → SREBP-2 down-regulation of LDLR → longer LDL residence time (all sizes).
But SFA (vs. carb) ↓ de-novo lipogenesis & TG synthesis → less CETP-mediated TG enrichment of LDL → fewer sd-LDL particles.
Net result: larger mean LDL size unless TG stays high.
5 Practical implications
Cutting SFA even further will not reliably shrink sd-LDL if your TG stay elevated.
Focus on:
lowering refined carbs / sugar,
soluble-fibre 10 g d⁻¹,
EPA-dominant omega-3 (2–4 g d⁻¹),
regular exercise.
Track apoB plus an NMR subclass panel; particle number matters more than size alone.
Bottom line
Saturated fat, by itself, is a weak lever on sd-LDL. Up to ~12–15 % of calories, it usually enlarges LDL particles when it displaces carbohydrate. Only when SFA intake is very high or coupled with elevated triglycerides (high-carb, insulin-resistant, or phenotype B) does small-dense LDL rise. If sd-LDL is your target, keep an eye on TG and apoB rather than obsessing over every extra gram of saturated fat. (BioMed Central, PMC, PMC, American Journal of Clinical Nutrition)
I haven’t seen any evidence of the LDL particle size hypothesis controlling for apoB in mendelian randomization studies, that should be the minimum criteria for establishing causation without confounding. The genes should be perceived to only affect LDL particle size but not apoB levels, which is hard to do.