Wonder why your HbA1C is too high or too low compared to your blood glucose levels?
I’m continuing my deep dives into the genetic pathways to get actionable insights as the previous ones have been incredible precise and useful. This time I’m looking at Glycation genetic pathways.
Here is the general description of the pathways and their variants. I will put the finding about my own genome below it as an example of what useful and actionable insights you can get.
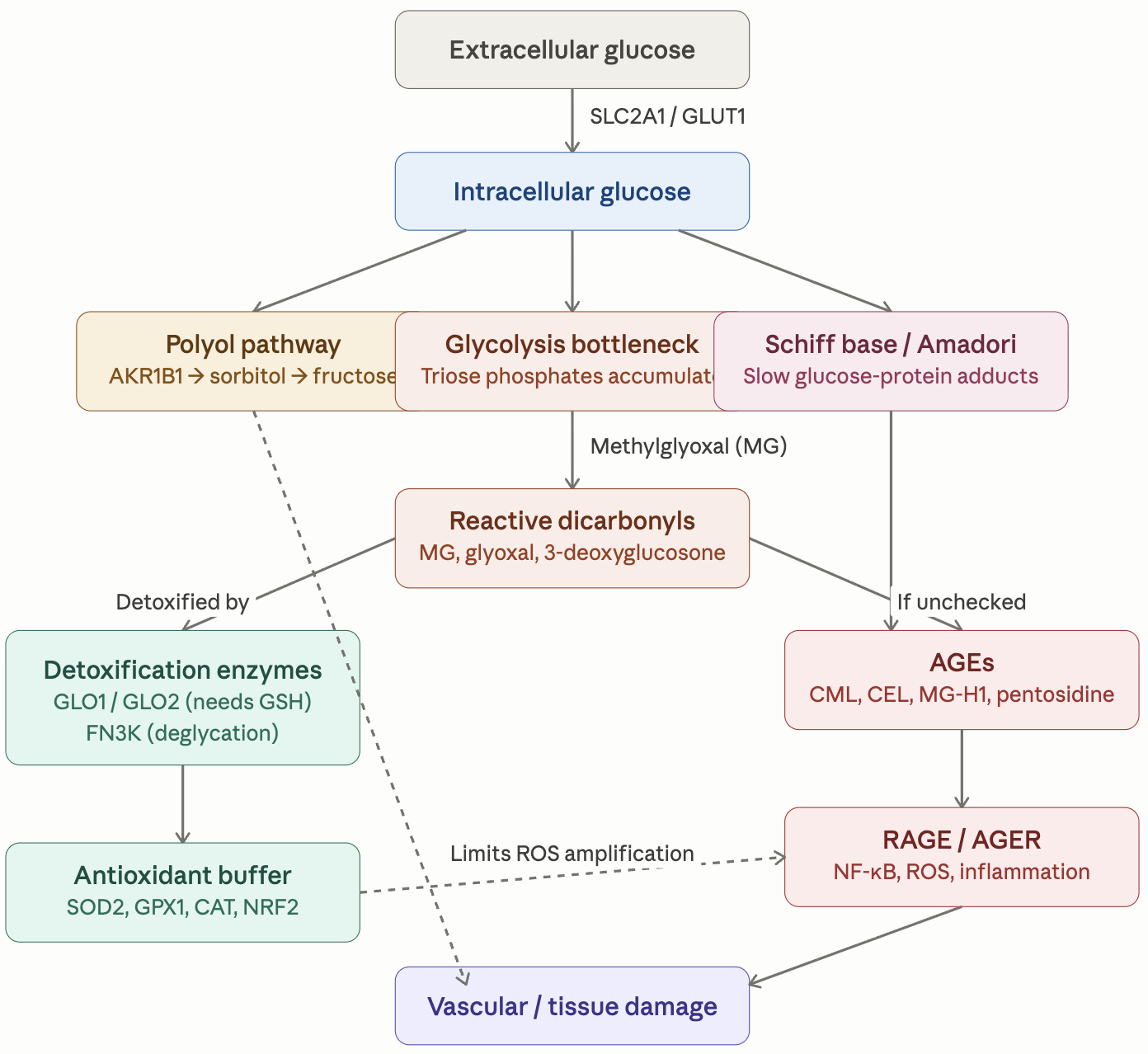
Glycation_Pathway_Reference.pdf (306.4 KB)
Here are the summary of the findings for my own genome. The full detailed report is 20 pages long.
Glycation Genetic Report — Top 10 Discussion Items
For your next physician appointment. Date: 2026-04-15. Source: personal WGS (60×, GRCh38). Companion: Glycation Genetic Report (full document).
The headline
Methylglyoxal-detoxification axis (GLO1 + NRF2-mediated GLO1 induction) is the dominant genetic vulnerability. Strongly favorable AGER (RAGE) profile partially offsets it. Existing diabetes and cardiovascular regimen is well-aligned. The two highest-priority gaps remain benfotiamine (upstream MG suppression) and sulforaphane (NRF2-driven GLO1 induction). L-carnosine is mechanistically near-ideal for the GLO1 bottleneck but is partially gated by your heterozygous CNDP1 genotype (intermediate carnosinase activity); it moves to a moderate-priority empirical trial rather than first-line.
Top 10 items to discuss
-
Add benfotiamine 150–300 mg/day. Lipid-soluble thiamine prodrug; activates transketolase, shunts triose phosphates away from methylglyoxal formation. Mechanistically targets the upstream side of the GLO1 bottleneck (Hammes et al., Nat Med 2003). Verify Momentous Multi thiamine content first — standard thiamine HCl is not bioequivalent. Not gated by any genotype you carry.
-
Add sulforaphane 10–40 mg/day (e.g., BroccoMax, Avmacol, MyrPro, or fresh broccoli sprouts). Most evidence-supported NRF2 activator; directly induces GLO1 transcription (Xue et al., Diabetes 2012). Addresses the three-SNP NRF2 stack identified in the report. Not gated by any genotype you carry.
-
L-carnosine — empirical 12-week trial, not lifetime commitment. Your CNDP1 is heterozygous at the lead Ahluwalia SNP (rs2346061) — intermediate carnosinase activity, not the (CTG)₅/₅ “Mannheim” strong-responder genotype. Recommended protocol if pursuing: 2 g/day split BID, co-time with NACET (which inhibits CN1 via S-cysteinylation), measure fructosamine and serum CML before/after, discontinue if no biomarker change. Mechanistically near-ideal for the GLO1 bottleneck (carnosine directly scavenges MG via its imidazole ring), but a meaningful fraction of each oral dose will be degraded in plasma before reaching kidney/endothelial tissue.
-
Verify Momentous Multi content. At half-dose, several cofactors may be sub-optimal: confirm actual delivered amounts of selenium (target 100–200 µg/day), manganese, thiamine, B6, and zinc before adding any of these separately.
-
Consider pyridoxamine 50–200 mg/day (a B6 form). Direct AGE-precursor scavenger; intercepts Amadori intermediates before progression to AGEs (Voziyan & Hudson 2005). Mechanistically complementary to FN3K and not gated by CNDP1 — a more reliable AGE-precursor scavenger than carnosine in your genotype context. Discuss US availability with physician.
-
Consider alpha-lipoic acid 300–600 mg/day (R-isomer preferred). Dual NRF2 activation and direct AGE-formation inhibition; also supports mitochondrial function (heterozygous SOD2 rs4880 finding). Insulin-sensitizing as a bonus.
-
AGER (RAGE) genetics are protective — don’t worry about RAGE inhibitors. The most replicated cardiometabolic SNP in this domain (rs2070600, GWAS p ≈ 10⁻⁵²) is wild-type. Soluble RAGE production is genetically intact. The clinical priority is upstream AGE limitation, not downstream RAGE blockade.
-
Empagliflozin and tirzepatide are well-aligned with the SLC2A1 + AKR1B1 + CNDP1 mesangial-vulnerability convergence. No change to the cardiometabolic regimen indicated by these findings — it’s already targeting the right bottleneck. The CNDP1 heterozygous finding adds a small increment to nephropathy-axis risk, reinforcing rather than changing the existing strategy.
-
Order baseline labs if not already recent: fasting + 2-hour postprandial glucose (or CGM if not in use), fructosamine (to compute glycation gap given heterozygous FN3K rs1056534), erythrocyte transketolase activation coefficient or plasma thiamine, total glutathione (GSH/GSSG ratio), serum selenium and zinc, plasma B6 (PLP), and skin autofluorescence (AGE Reader) if available. If pursuing the carnosine trial: add serum CN1 activity measurement (Mannheim methodology / commercial ELISA) and paired serum CML or MG-H1 before and after the 12-week trial.
-
Ongoing monitoring: HbA1c every 3–6 months, UACR + eGFR annually, dilated retinal exam annually, hsCRP annually. Particular attention to time-in-range (CGM) and glycemic variability rather than HbA1c alone — postprandial spikes drive MG generation more than mean glucose does.
Quick reference — primary genetic findings
| Finding | Status | Strength of evidence |
|---|---|---|
| GLO1 rs1049346 AA | Homozygous risk | Strong (Peculis 2013, p = 2.6 × 10⁻⁵) |
| GLO1 rs1130534 + rs4746 | Both heterozygous | Moderate; additive |
| NFE2L2 rs6721961 + rs35652124 + rs2706110 | Two homozygous, one het | Moderate; cumulative |
| AKR1B1 rs759853 TT | Homozygous | Direction context-dependent |
| SLC2A1 rs841853 + rs841847 | Both homozygous (LD) | Moderate (DN meta-analyses) |
| SOD2 rs4880 AG | Heterozygous Val/Ala | Moderate |
| CNDP1 rs2346061 CA | Heterozygous (intermediate CN1) | Moderate (Ahluwalia 2011, p = 5.07 × 10⁻⁴) |
| AGER rs2070600 | Wild-type Gly/Gly | Very strong protective (GWAS p ≈ 10⁻⁵²) |
| FN3K rs3859206 AA | Homozygous favorable | Moderate protective |
| MMP1 rs1799750 het | Mildly favorable | Moderate |
All variants high-confidence calls (DP 30–110, GQ 99, MQ 60). AGER variants not detected at 60× WGS = high-confidence absence-of-call. CNDP1 (CTG)ₙ leucine repeat itself indeterminate by short-read WGS; tag-SNP haplotype inference points to intermediate (not Mannheim 5L/5L) phenotype.
Quick reference — supplement priority for glycation pathway
| Priority | Intervention | Genotype-gated? |
|---|---|---|
| HIGH | Benfotiamine 150–300 mg/day | No |
| HIGH | Sulforaphane 10–40 mg/day | No |
| MODERATE | Pyridoxamine 50–200 mg/day | No |
| MODERATE | Alpha-lipoic acid 300–600 mg/day | No |
| MODERATE — empirical trial | L-carnosine 2 g/day BID + NACET co-timing | Yes — CNDP1 heterozygous (intermediate response predicted) |
| WATCHLIST | Carnosinol / carnostatine + carnosine | Future — designed to bypass CN1 gate |