About my sleep: when I was taking melatonin, 3 - 5 mg my sleep was ok (7-8 h), when I stopped it’s still ok but the deep sleep stage increased. Coincidence?
It’s well studied that 5 mg is worse for sleep than 0.3 mg - 1 mg.
Have you searched for any clinical trials or mendelian randomization studies supporting this idea?
1 Like
1 Like
Interesting study comparing BAM15 to atorvastatin.
"In summary, the in vivo experiments demonstrated that oral administration of BAM15 significantly reduced the increased plaque area and the increased necrotic core area of plaque induced by WD feeding in ApoE(−/−) mice. In in vitro experiments, cell biological assays confirmed that BAM15 inhibited RAW264.7 macrophages invasive ability and reduced PA-induced lipid accumulation. By combining RNA-seq techniques, molecular docking, and experimental validation, we found that IL-1α, SRC, and CSF3 are key anti-AS molecules of BAM15.
This study reveals that BAM15 may be a promising drug against AS, providing scientific evidence to reveal the pharmacological mechanism of BAM15 in the treatment of AS, and facilitating future clinical translational research."
Combining RNA-seq, molecular docking and experimental verification to explore the mechanism of BAM15 as a potential drug for atherosclerosis:
https://www.nature.com/articles/s41598-025-98209-3
2 Likes
The practical question is what’s causing what. Clinical trials and mendelian randomization studies that separately investigate a change in either apoB or apoA-1 levels tells us whether it’s higher apoB or lower apoA-1 that’s increasing the risk, or vice versa lower apoB or higher apoA-1 that is decreasing the risk, if that makes sense.
So correct me if I’m wrong if you believe that the ratio is a better predictor than apoB you would need to know whether apoA-1 independently changes risk of apoB levels, as that would tell you whether a lower ratio from higher apoA-1 levels would decrease risk.
I think this is wrong: Melatonin megadoses? - #316 by adssx
Also, let’s stick to cardio health here. If you want to discuss melatonin, please answer in the above thread.
4 Likes
Atherosclerotic blood vessel cells grow similar to tumors, study reveals
"Researchers from the University of Southern Denmark and Odense University Hospital have studied tissue from patients with atherosclerosis. They found that many of the cells in the diseased tissue carried the same genetic alteration and appeared to originate from a single ancestral cell that had divided repeatedly—a pattern otherwise associated with tumor biology.
In several patients, a large proportion of the cells were derived from one single mutated cell that had undergone many rounds of cell division.
“It’s striking how many cells in the tissue share the exact same genetic change. In several samples, more than 10% of the cells—hundreds of thousands cells—carried the same alteration. It’s difficult to interpret this as anything other than all these cells originating from a shared ancestral cell that, at some point during disease development, acquired the mutation,” says Lasse Bach Steffensen, Associate Professor at the Department of Molecular Medicine at the University of Southern Denmark.
In a tumor, the disease often begins when a single cell acquires a genetic alteration that causes it to divide more than it should. As the daughter cells inherit the same alteration and continue to divide, a progressively larger mass of cells forms—what we call a tumor.
This finding offers a new perspective on atherosclerosis—a disease primarily associated with cholesterol, inflammation and lifestyle factors."
6 Likes
Small, associative study…
3 Likes
They found higher magnesium implies lower CIMT, but also higher haemoglobin implies lower CIMT.
2 Likes
Here’s evidence that dietary cholesterol increases serum cholesterol, so it’s not bunk at all that it does, it depends on how much dietary cholesterol you eat at baseline.
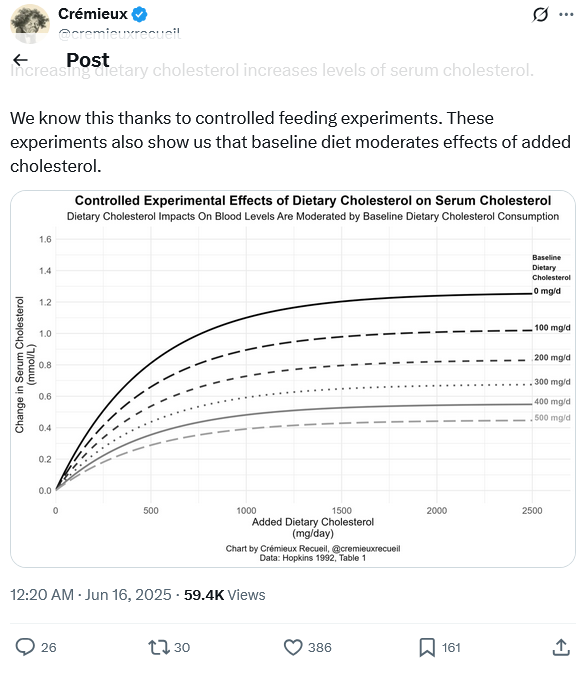
6 Likes
It’s eggover. Where am I supposed to get my choline now?
3 Likes
Lecithin works for people if you really want it… ![]()
3 Likes
If you are on ezetimibe, and even statin, does dietary cholesterol matter? I might find out, as I consume 2-3 eggs weekly, and have added 10mg/day of ezetimibe to my 4mg/day pitavastatin, will get a lipid panel early August.
4 Likes
I have to say I’ve tried different dosing of melatonin and 0.3-0.5mg works just as well for sleeping for me as higher doses. Maybe even better.
3 Likes
There is a reference to a Hopkins, 1992 article, a single one. Shouldn’t we consider multiple papers, especially if there are more recent ones? I do not exclude that the one indicated is the seminal paper which has not been contradicted, but how do we know?
2 Likes
OK, it’s available in PDF format, but again, I woudl like to know if tghe findings have been confirmed by more recent studies or not.

The Deepseek AI seems to provide some hints, maybe we should include the recent literature in our considerations
The 1992 Hopkins meta-analysis (Am J Clin Nutr. 1992;55:1060-1070) provides critical insights into how baseline dietary cholesterol consumption influences serum cholesterol responses. Here’s a detailed breakdown of its findings on individual variability:
1. Baseline Dietary Cholesterol Intake Matters
-
Adaptive Downregulation:
Individuals with habitually high dietary cholesterol intake (e.g., >400 mg/day) showed minimal changes in serum cholesterol when challenged with additional cholesterol. Their bodies compensated by reducing endogenous cholesterol synthesis and increasing excretion. -
Low-Baseline Hyper-Response:
Subjects with low habitual intake (e.g., <200 mg/day) experienced significantly greater serum cholesterol increases when dietary cholesterol rose. Their regulatory systems were less “primed” to adapt.
2. Individual Variation in Serum Response
The study identified three key variables explaining response differences:
A. Genetic Factors
- APOE4 carriers: Elevated LDL-C response (up to 2× higher than non-carriers).
- APOE2 carriers: Blunted response (even LDL-C decreases in some).
B. Baseline Serum Cholesterol Levels
- People with low baseline LDL-C (<100 mg/dL) showed greater absolute increases than those with high baseline levels.
C. Metabolic Health
- Insulin resistance, obesity, and diabetes amplified cholesterol absorption.
3. Quantitative Effects
-
Average Response:
Every 100 mg added dietary cholesterol/day → ~2.2 mg/dL increase in total serum cholesterol.- LDL-C: Increased by ~1.9 mg/dL per 100 mg.
- HDL-C: Minor rise (~0.4 mg/dL).
-
Hyper-Responders:
Experienced up to 4–6 mg/dL LDL-C increases per 100 mg of added cholesterol.
4. Interaction with Saturated Fat
Dietary cholesterol’s impact was dwarfed by saturated fat:
- Saturated fat (per 1% energy increase) → LDL-C ↑ ~2.4 mg/dL.
-
Dietary cholesterol (per 100 mg/day) → LDL-C ↑ ~1.9 mg/dL.
→ Reducing saturated fat was 5–6× more effective for lowering LDL than restricting dietary cholesterol.
5. Practical Implications
-
“Cholesterol Tolerance”:
Habitual egg consumers may see no LDL increase from additional eggs, while new consumers might. -
Clinical Guidance:
- General population: No need to restrict cholesterol if saturated fat is low.
- Hyper-responders (or APOE4+): Limit cholesterol-rich foods (egg yolks, organ meats).
Key Quote from the Study:
“The response to dietary cholesterol is highly variable and depends on an individual’s adaptive metabolic state, genetics, and background diet.”
Modern Context
Later studies (e.g., JAMA, 2019; Nutrients, 2020) confirmed:
- Egg consumption (1–3/day) rarely affects LDL in most people.
- Whole-food matrix effects: Eggs’ phospholipids and antioxidants may mitigate cholesterol absorption.
- Guidelines now focus on reducing saturated fats and ultra-processed foods over dietary cholesterol.
For personalized advice, lipid testing pre/post dietary changes remains gold-standard.
4 Likes
Have you found any that stratified by baseline dietary cholesterol intake?
NO, even though I did not search. Do you think that’s an actionable aspect? What discouraged me from elaborating further on that graph is that in the article, fig. 3, for a determined subset of population (LDL <= 50 mg/dL), the variability is huge. Even though that is a rare subgroup, we know nothing about the variability of other subgroups and this makes the graph of figure 2, based on average trends, probably not so useful in practice. Maybe thsi is why the more recent studies focused on genetic variability, that apparently governs the synthesis and the absorption of cholesterol. Also, it seems that usually the intake of saturated fats governs over the intake of choelsterol, but this may be true only in determined conditions. At the end, the graph displays one average aspect of a very complex system, where apparently genetics governs.
My conclusions are that, if we want to decide whether to eat eggs or not and how many, every single individual should keep fixed the intake of saturated fats, their lifestyle, then measure the baseline cholesterol without eggs, then take measures after adding various amounts of eggs. Probably it’s too much of a laborious task, so some simplifications are needed.
1 Like
Doesn’t that mean you don’t believe any average result is applicable since you don’t know what part of the dose-efficacy curve you are on, without verifying for yourself and don’t expect the average response until further evidence? (e.g for drugs).
I don’t disagree that you could test whether LDL-C lowers for you before deciding to lower dietary cholesterol intake if that’s important to you. You need to know how long you need to test for and as N=1 it can be confounded.
But regardless this should clear up the assumption that it’s not possible.