Thanks for your analysis and insight. I looked around a little and came across a few things that Shift Bioscience, the company doing the research, had to say.
They did find function:
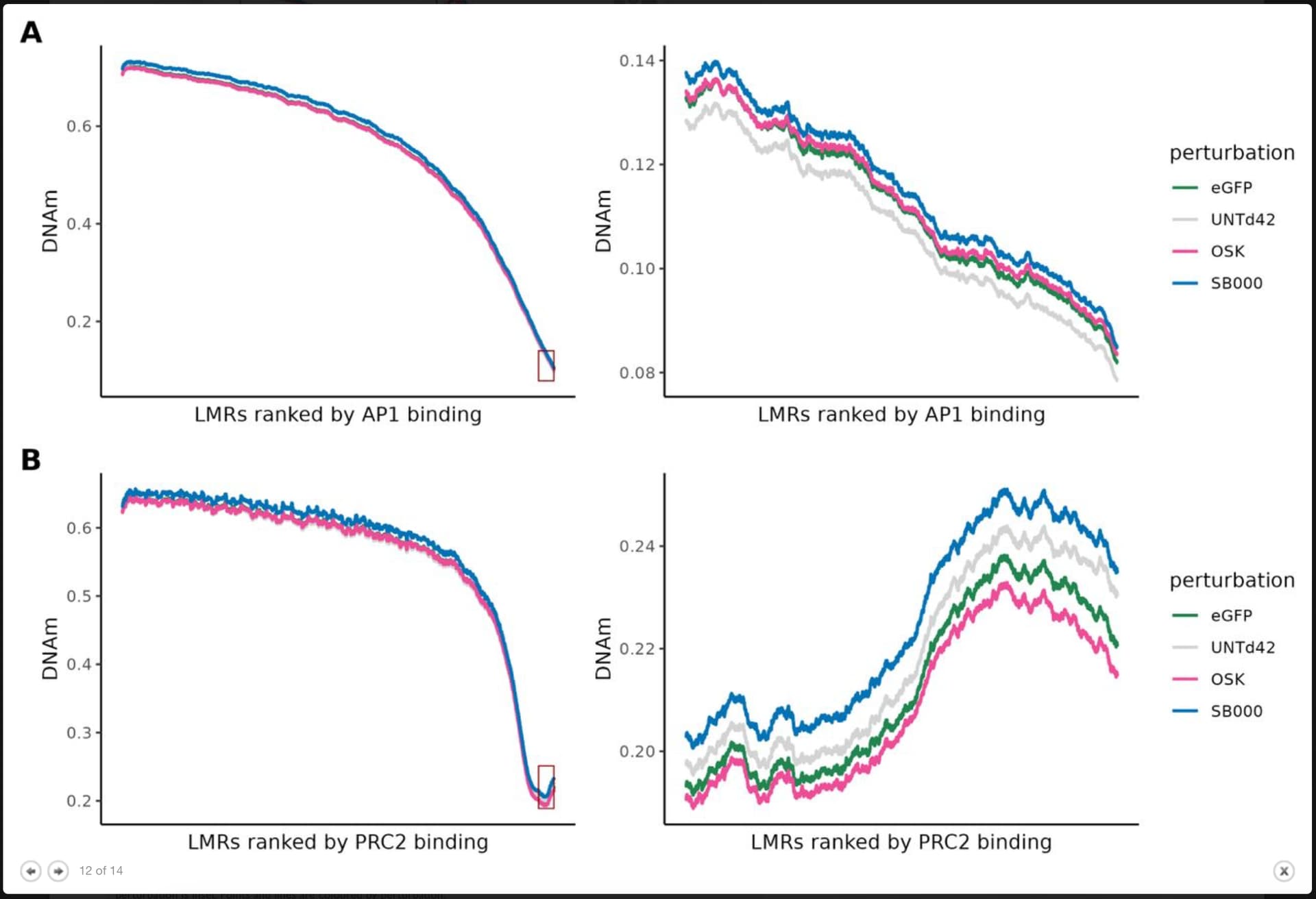
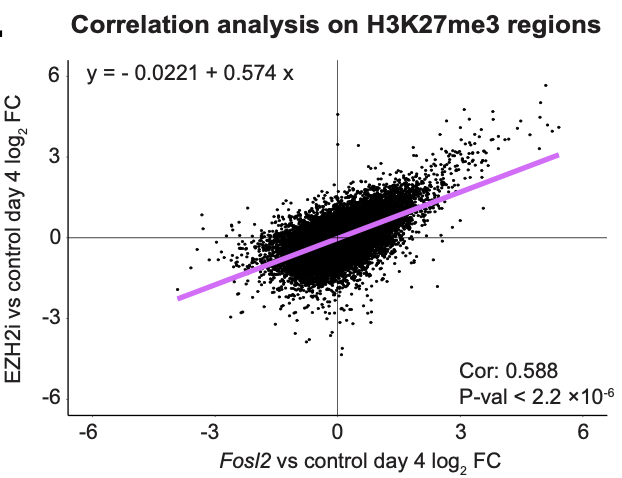
SB000’s functional effects – improved collagen production and preserved fibroblast behavior – suggest real potential for reversing tissue aging.
Immediate plans:
With these promising results, Shift is moving quickly to validate SB000 further. The next stage involves:
• Testing across an expanded range of disease-relevant human cell types
• Investigating whether rejuvenated cells functionally behave like their younger counterparts
• Launching proof-of-concept in vivo studies, including in mouse models
From a 2022 interview with Daniel Ives, a co-founder of the company, the expectation was that it was almost certainly going to be a therapy with multiple genes, not the single gene they ended up finding. It has been noted that regulatory approval will be easier than it would be with multiple factors.
The interview with Longevity Technology, done for the purpose of raising funds, is sort of long, but if this development has captured your interest as much as it has mine, time will be suspended:
Using a novel machine learning approach, British startup Shift Bioscience is on the verge of validating drug targets for cellular reprogramming. The company is currently in the process of raising £3.5 million to begin the target validation phase for genes that could become the targets of rejuvenation therapies.
Longevity.Technology: Cellular rejuvenation holds great potential but it is also fraught with challenges. With significant money now flowing into the rejuvenation field, Shift Bioscience is a player with a unique position in this hot investment area: the ability to identify safe targets. We caught up with co-founder and CEO Dr Daniel Ives to learn more.
Ives has been working in the field of aging since 2009, and focused on mitochondrial rejuvenation at the University of Cambridge and the Crick Institute. He co-founded Shift in 2017, initially with the aim of developing mitochondrial drugs for age-related diseases, but then a surprise discovery put the company on a different path.
“What we decided to do was to attempt a CRISPR screen for aging, which would allow us to disrupt the function of any gene across 20,000 genes, and then the data would tell us what’s the best point of leverage,” explains Ives. “To do this, we used machine learning to create a single cell aging clock built out of genes, not out of methylation sites. And we were about to go ahead and do the CRISPR screen, when we realised that we had done a lot more than just create a clock.”
While methylation clocks provide very little information, biologically, about what aging is linked to, Shift’s clock was based on genes, which Ives points out are very rich in data.
“Genes have annotation, there’s a lot of information linked to them, so when we looked at our clock, which is made out of genes, there were mitochondrial genes, and there were ribosomal genes, and there was even a gene that was sufficient to accelerate aging on its own,” he says. “And so we realised that our clock was not just providing a readout of aging, it was a window into aging.”
The discovery thrilled the team at Shift because it provided an unprecedented view of aging biology that was also completely unbiased.
“It’s completely open because it includes known biology, but it also includes unknown biology, so you’re not blinkered by what you know,” says Ives. “To the extent you can measure things, you can ask the universe what is linked to aging, and you get a collection of genes in an unbiased way.”
Identifying safe genes
The second key thing that Shift realised was that doing a CRISPR screen, as the team had originally planned, was a constrained approach, because it would only disrupt gene functions one by one.
“There might be a way to change aging with one gene, but there’s a strong possibility there isn’t,” says Ives, citing pluripotency, where stem cells are created from normal cells using four factors, not just one.
“So we’ll use a combinatorial approach, where we throw all these genes we’ve identified into cells, capture the cells that rejuvenate, then figure out what single genes or gene combinations drove those cells.”
But perhaps the most important thing to come out of Shift’s approach is the identification of safe cellular reprogramming targets. While cellular reprogramming has been measured by DNA methylation clocks to be one of the most powerful potential interventions against aging, its key challenge to date has been safety.
“What we decided to do was to attempt a CRISPR screen for aging, which would allow us to disrupt the function of any gene across 20,000 genes, and then the data would tell us what’s the best point of leverage,” explains Ives. “To do this, we used machine learning to create a single cell aging clock built out of genes, not out of methylation sites. And we were about to go ahead and do the CRISPR screen, when we realised that we had done a lot more than just create a clock.”
A window into aging
While methylation clocks provide very little information, biologically, about what aging is linked to, Shift’s clock was based on genes, which Ives points out are very rich in data.
“Genes have annotation, there’s a lot of information linked to them, so when we looked at our clock, which is made out of genes, there were mitochondrial genes, and there were ribosomal genes, and there was even a gene that was sufficient to accelerate aging on its own,” he says. “And so we realised that our clock was not just providing a readout of aging, it was a window into aging.”
The discovery thrilled the team at Shift because it provided an unprecedented view of aging biology that was also completely unbiased.
“It’s completely open because it includes known biology, but it also includes unknown biology, so you’re not blinkered by what you know,” says Ives. “To the extent you can measure things, you can ask the universe what is linked to aging, and you get a collection of genes in an unbiased way.”
Identifying safe genes
The second key thing that Shift realised was that doing a CRISPR screen, as the team had originally planned, was a constrained approach, because it would only disrupt gene functions one by one.
“There might be a way to change aging with one gene, but there’s a strong possibility there isn’t,” says Ives, citing pluripotency, where stem cells are created from normal cells using four factors, not just one. “So we’ll use a combinatorial approach, where we throw all these genes we’ve identified into cells, capture the cells that rejuvenate, then figure out what single genes or gene combinations drove those cells.”
But perhaps the most important thing to come out of Shift’s approach is the identification of safe cellular reprogramming targets. While cellular reprogramming has been measured by DNA methylation clocks to be one of the most powerful potential interventions against aging, its key challenge to date has been safety.
“What we decided to do was to attempt a CRISPR screen for aging, which would allow us to disrupt the function of any gene across 20,000 genes, and then the data would tell us what’s the best point of leverage,” explains Ives. “To do this, we used machine learning to create a single cell aging clock built out of genes, not out of methylation sites. And we were about to go ahead and do the CRISPR screen, when we realised that we had done a lot more than just create a clock.”
“The whole problem around reprogramming has been that it’s a Goldilocks approach – too little and you get nothing, but too much and it’s cancer,” says Ives. “It’s an existential problem for a reprogramming therapy – it only takes one premalignant cell in your body to be tipped into malignancy and that’s the beginning of the cancer.”
“We’ve used our approach to predict the genes driving rejuvenation during reprogramming – they don’t look cancerous and our next step is to throw them into a system to see if we can rejuvenate safely. We can show this definitively – we can sort the cells for rejuvenation and for pluripotency, and then we can pinpoint those genes which not only rejuvenate but are also safe.”
De-risking rejuvenation
To complete this pivotal next step, Shift needs to refuel with new funds. Everything to date has been conducted in silico and the company is eager to test its gene predictions in the lab, firstly to align its machine learning systems with what happens in the dish.
“When we get into the dish, it is going to tell us how to improve our bioinformatics until it is as good as it can be,” says Ives. “And then we’ll start patenting based on the genes it identifies, which will include mRNA therapeutics targeting those genes, as well drug targets on the gene products. And that’s the basis for a fully de-risked rejuvenation therapy.
“It’s the best possible foundation for preclinical and clinical development, because you’ve first asked what’s the best biology and then you start where the data tells you to start. We don’t know exactly what we’re going to get up front, it might be a combination of two or three genes, or it could be up to five, but that will dictate what the therapy is going to look like. It’s almost certainly going to be a therapy targeting multiple genes, which will require an innovative approach, but if this is what the biology tells us, we’ve got to listen.”
Ives expects the new funding round will support Shift through the next couple of years, taking the company through the identification of a spectrum of safe rejuvenation genes, narrowing down to a preferred set of genes, then patenting the therapeutic options around those genes.